

Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place
- PMID: 29653955
- PMCID: PMC5935233
- DOI: 10.1158/2159-8290.CD-17-1461
Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place
Abstract
Oncogene activation disturbs cellular processes and accommodates a complex landscape of changes in the genome that contribute to genomic instability, which accelerates mutation rates and promotes tumorigenesis. Part of this cellular turmoil involves deregulation of physiologic DNA replication, widely described as replication stress. Oncogene-induced replication stress is an early driver of genomic instability and is attributed to a plethora of factors, most notably aberrant origin firing, replication-transcription collisions, reactive oxygen species, and defective nucleotide metabolism.Significance: Replication stress is a fundamental step and an early driver of tumorigenesis and has been associated with many activated oncogenes. Deciphering the mechanisms that contribute to the replication stress response may provide new avenues for targeted cancer treatment. In this review, we discuss the latest findings on the DNA replication stress response and examine the various mechanisms through which activated oncogenes induce replication stress. Cancer Discov; 8(5); 537-55. ©2018 AACR.
©2018 American Association for Cancer Research.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
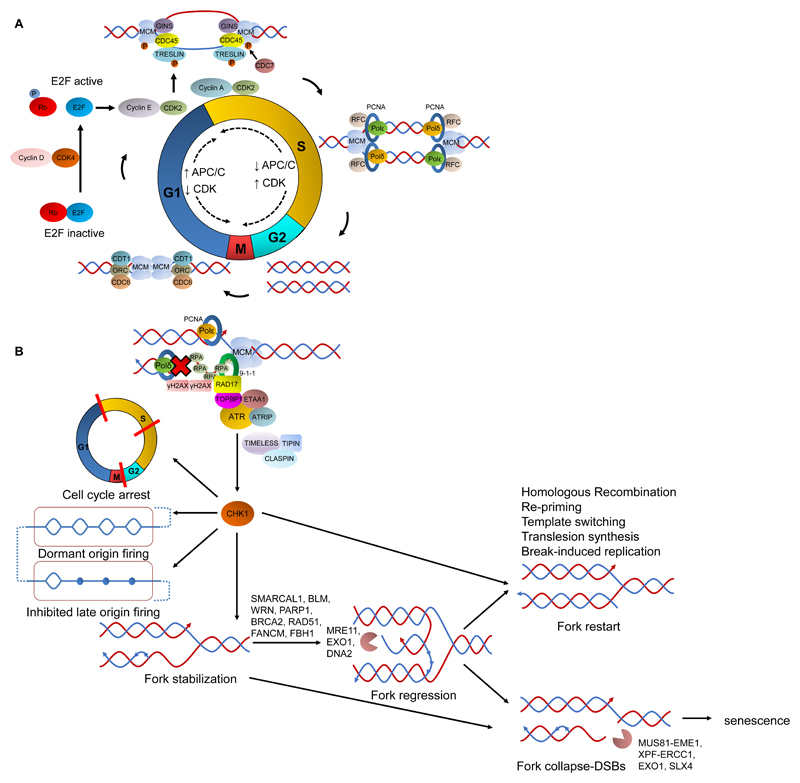
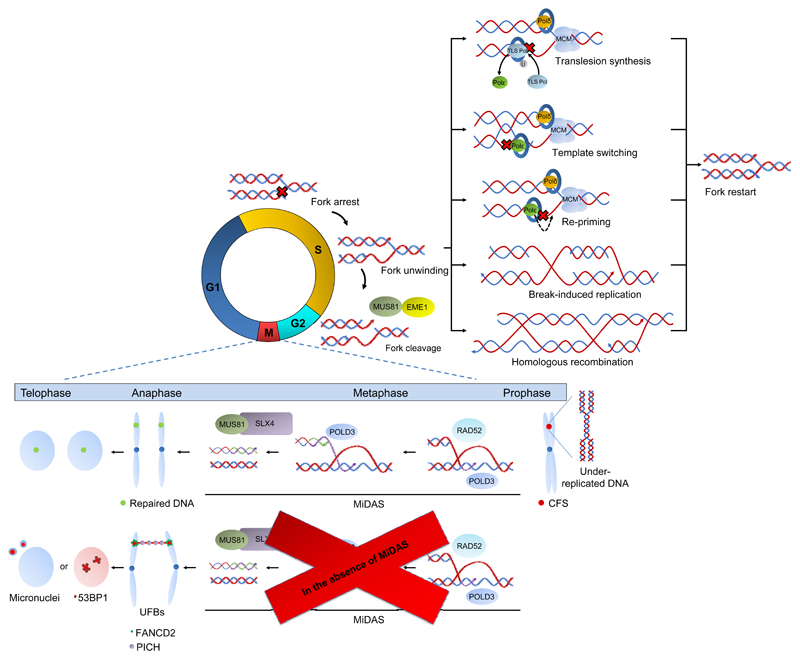
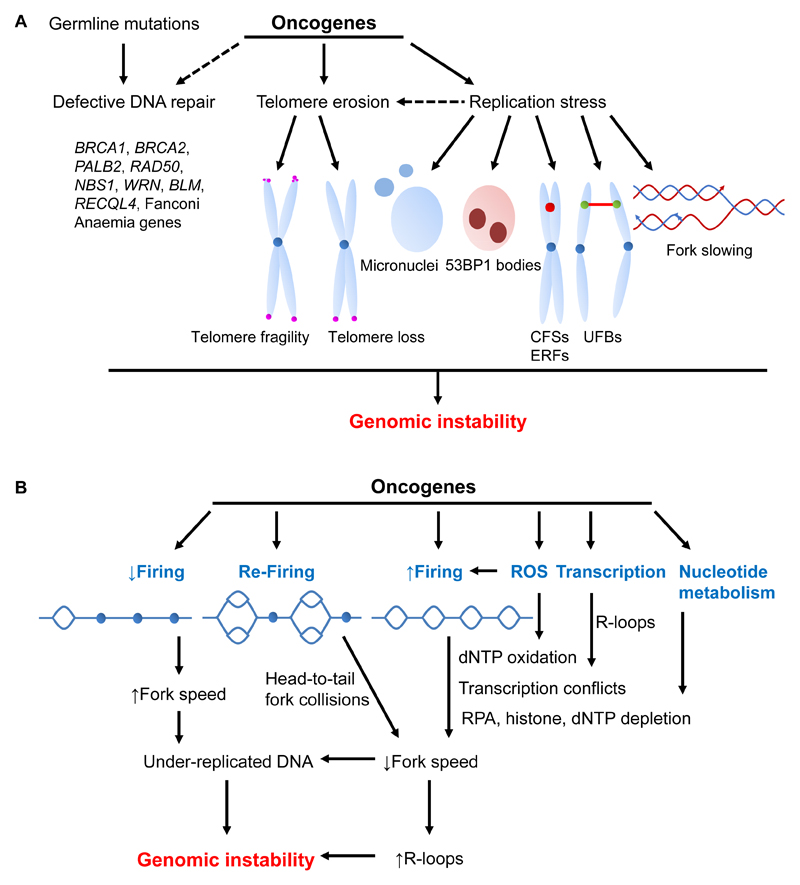
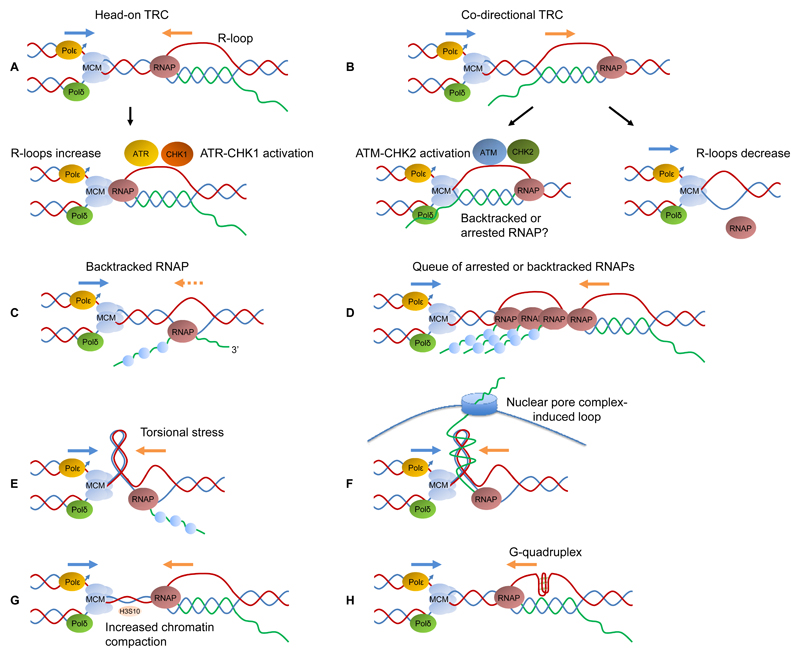

References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. - PubMed
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. - PubMed
-
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
