

Glycine Amidinotransferase (GATM), Renal Fanconi Syndrome, and Kidney Failure
- PMID: 29654216
- PMCID: PMC6050927
- DOI: 10.1681/ASN.2017111179
Glycine Amidinotransferase (GATM), Renal Fanconi Syndrome, and Kidney Failure
Abstract
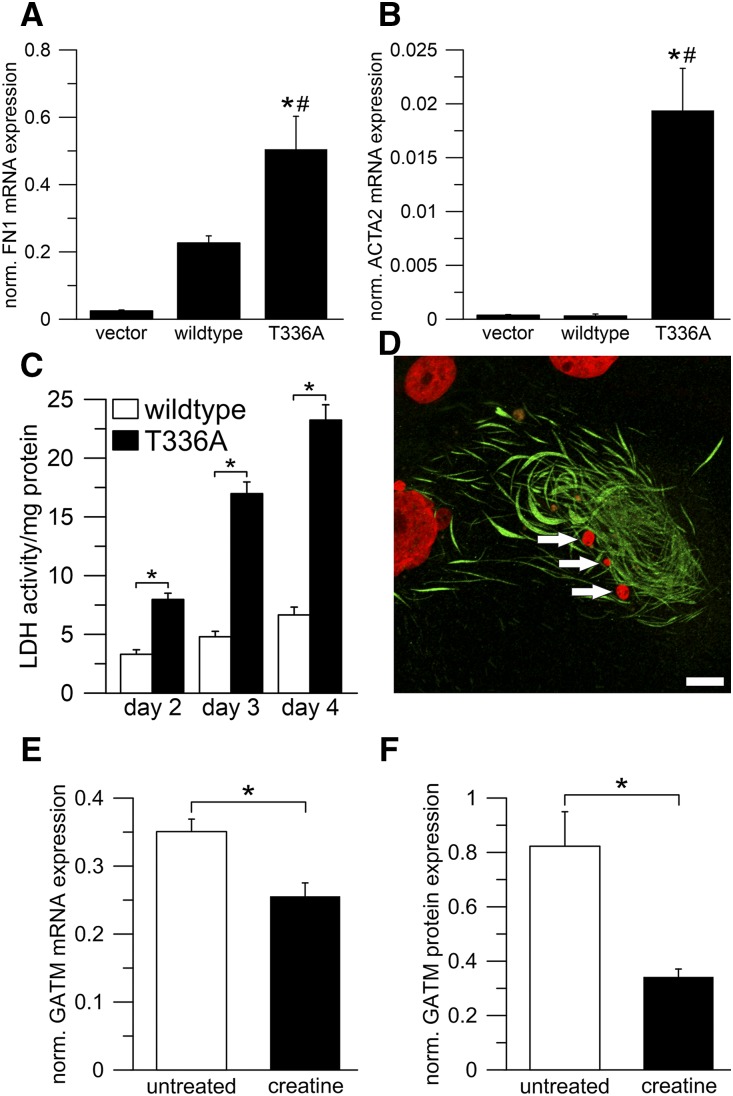
Background For many patients with kidney failure, the cause and underlying defect remain unknown. Here, we describe a novel mechanism of a genetic order characterized by renal Fanconi syndrome and kidney failure.Methods We clinically and genetically characterized members of five families with autosomal dominant renal Fanconi syndrome and kidney failure. We performed genome-wide linkage analysis, sequencing, and expression studies in kidney biopsy specimens and renal cells along with knockout mouse studies and evaluations of mitochondrial morphology and function. Structural studies examined the effects of recognized mutations.Results The renal disease in these patients resulted from monoallelic mutations in the gene encoding glycine amidinotransferase (GATM), a renal proximal tubular enzyme in the creatine biosynthetic pathway that is otherwise associated with a recessive disorder of creatine deficiency. In silico analysis showed that the particular GATM mutations, identified in 28 members of the five families, create an additional interaction interface within the GATM protein and likely cause the linear aggregation of GATM observed in patient biopsy specimens and cultured proximal tubule cells. GATM aggregates-containing mitochondria were elongated and associated with increased ROS production, activation of the NLRP3 inflammasome, enhanced expression of the profibrotic cytokine IL-18, and increased cell death.Conclusions In this novel genetic disorder, fully penetrant heterozygous missense mutations in GATM trigger intramitochondrial fibrillary deposition of GATM and lead to elongated and abnormal mitochondria. We speculate that this renal proximal tubular mitochondrial pathology initiates a response from the inflammasome, with subsequent development of kidney fibrosis.
Keywords: AGAT; fibrosis; mitochondriopathy; protein deposits; tubulopathy.
Copyright © 2018 by the American Society of Nephrology.
Figures
Comment in
-
GATM mutations cause mitochondrial abnormalities and kidney failure.Nat Rev Nephrol. 2018 Jul;14(7):414. doi: 10.1038/s41581-018-0017-3. Nat Rev Nephrol. 2018. PMID: 29695752 No abstract available.
-
GATM Mutations Cause a Dominant Fibrillar Conformational Disease in Mitochondria-When Eternity Kills.J Am Soc Nephrol. 2018 Jul;29(7):1787-1789. doi: 10.1681/ASN.2018040450. Epub 2018 May 22. J Am Soc Nephrol. 2018. PMID: 29789432 Free PMC article. No abstract available.
-
Genetic mitochondrial glycine amidinotransferase protein aggregate formation triggers microparticle sensing and kidney failure.Ann Transl Med. 2018 Aug;6(16):315. doi: 10.21037/atm.2018.07.09. Ann Transl Med. 2018. PMID: 30364060 Free PMC article. No abstract available.
References
-
- Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al.: Chronic kidney disease: Global dimension and perspectives. Lancet 382: 260–272, 2013 - PubMed
-
- Dent CE, Harris H: The genetics of cystinuria. Ann Eugen 16: 60–87, 1951 - PubMed
-
- Smith R, Lindenbaum RH, Walton RJ: Hypophosphataemic osteomalacia and Fanconi syndrome of adult onset with dominant inheritance. Possible relationship with diabetes mellitus. Q J Med 45: 387–400, 1976 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
