

A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis
- PMID: 29656858
- PMCID: PMC5986694
- DOI: 10.1016/j.ajhg.2018.03.005
A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis
Erratum in
-
A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis.Am J Hum Genet. 2018 Oct 4;103(4):631. doi: 10.1016/j.ajhg.2018.09.002. Am J Hum Genet. 2018. PMID: 30290155 Free PMC article. No abstract available.
Abstract
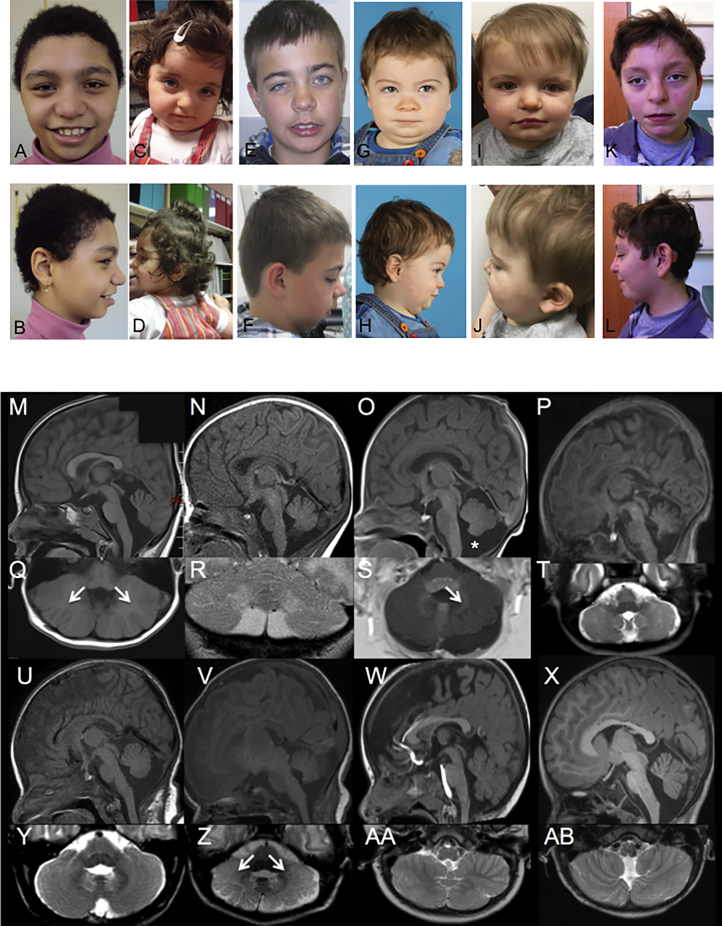
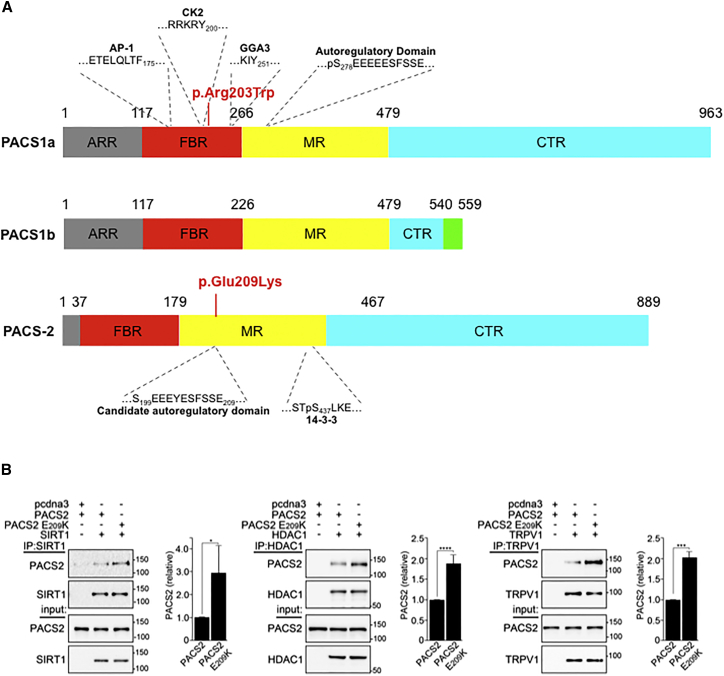
Developmental and epileptic encephalopathies (DEEs) represent a large clinical and genetic heterogeneous group of neurodevelopmental diseases. The identification of pathogenic genetic variants in DEEs remains crucial for deciphering this complex group and for accurately caring for affected individuals (clinical diagnosis, genetic counseling, impacting medical, precision therapy, clinical trials, etc.). Whole-exome sequencing and intensive data sharing identified a recurrent de novo PACS2 heterozygous missense variant in 14 unrelated individuals. Their phenotype was characterized by epilepsy, global developmental delay with or without autism, common cerebellar dysgenesis, and facial dysmorphism. Mixed focal and generalized epilepsy occurred in the neonatal period, controlled with difficulty in the first year, but many improved in early childhood. PACS2 is an important PACS1 paralog and encodes a multifunctional sorting protein involved in nuclear gene expression and pathway traffic regulation. Both proteins harbor cargo(furin)-binding regions (FBRs) that bind cargo proteins, sorting adaptors, and cellular kinase. Compared to the defined PACS1 recurrent variant series, individuals with PACS2 variant have more consistently neonatal/early-infantile-onset epilepsy that can be challenging to control. Cerebellar abnormalities may be similar but PACS2 individuals exhibit a pattern of clear dysgenesis ranging from mild to severe. Functional studies demonstrated that the PACS2 recurrent variant reduces the ability of the predicted autoregulatory domain to modulate the interaction between the PACS2 FBR and client proteins, which may disturb cellular function. These findings support the causality of this recurrent de novo PACS2 heterozygous missense in DEEs with facial dysmorphim and cerebellar dysgenesis.
Keywords: PACS-2; PACS2; cerebellar dysgenesis; epilepsy; intellectual disability.
Copyright © 2018 American Society of Human Genetics. All rights reserved.
Figures
References
-
- Eltze C.M., Chong W.K., Cox T., Whitney A., Cortina-Borja M., Chin R.F., Scott R.C., Cross J.H. A population-based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54:437–445. - PubMed
-
- von Deimling M., Helbig I., Marsh E.D. Epileptic encephalopathies-clinical syndromes and pathophysiological concepts. Curr. Neurol. Neurosci. Rep. 2017;17:10. - PubMed
-
- Tavyev Asher Y.J., Scaglia F. Molecular bases and clinical spectrum of early infantile epileptic encephalopathies. Eur. J. Med. Genet. 2012;55:299–306. - PubMed
-
- McTague A., Howell K.B., Cross J.H., Kurian M.A., Scheffer I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–316. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
