

Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal
- PMID: 29656891
- PMCID: PMC5927631
- DOI: 10.1016/j.cell.2018.02.020
Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal
Abstract
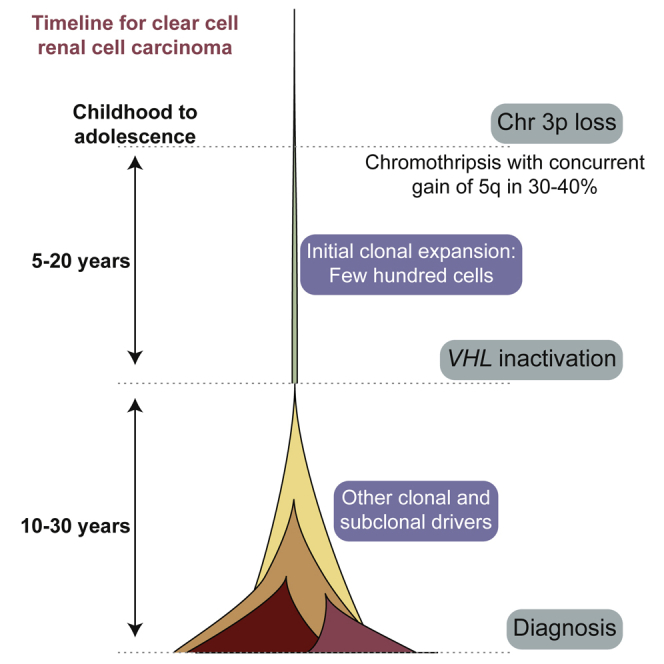
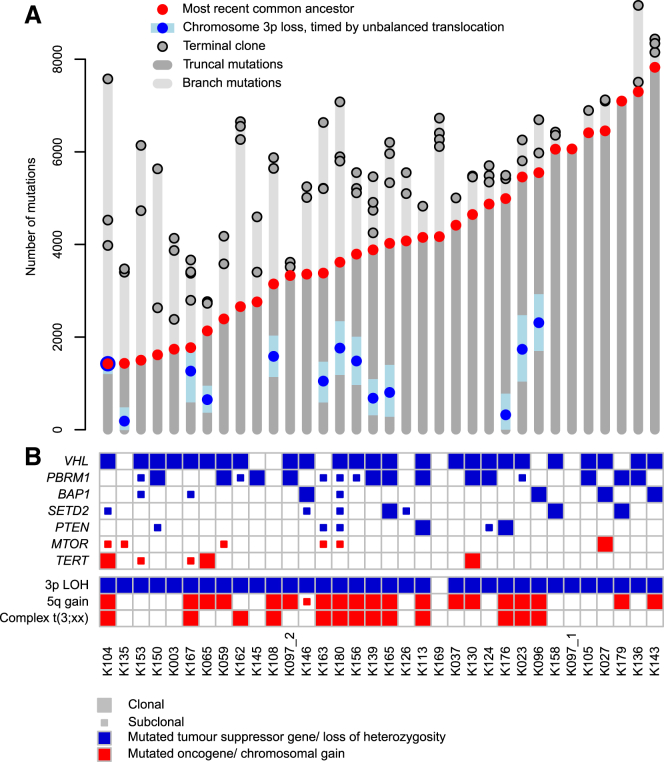
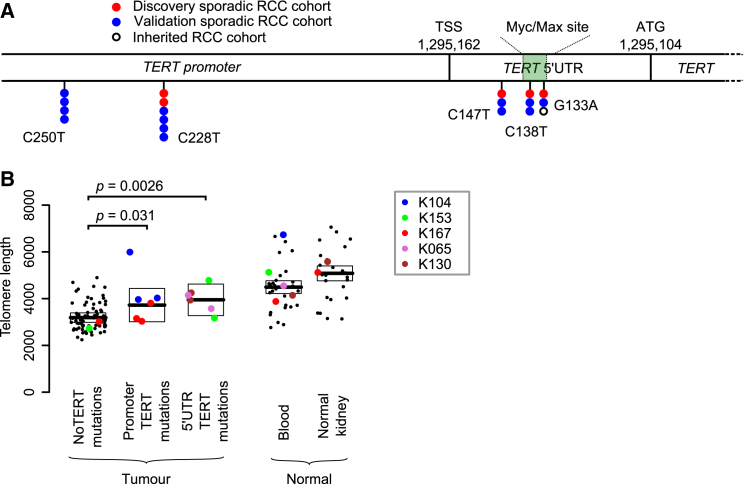
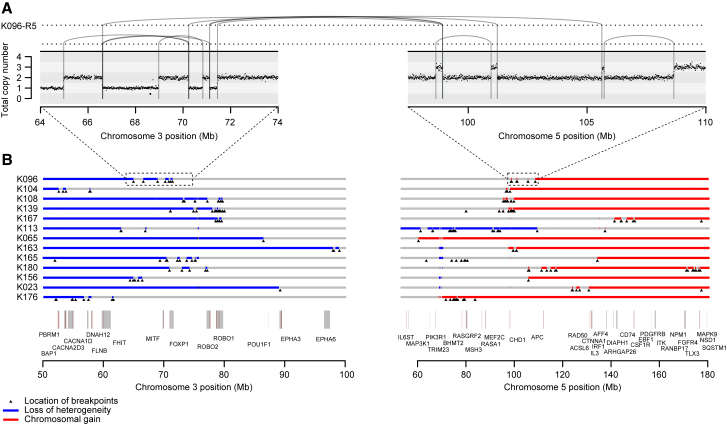
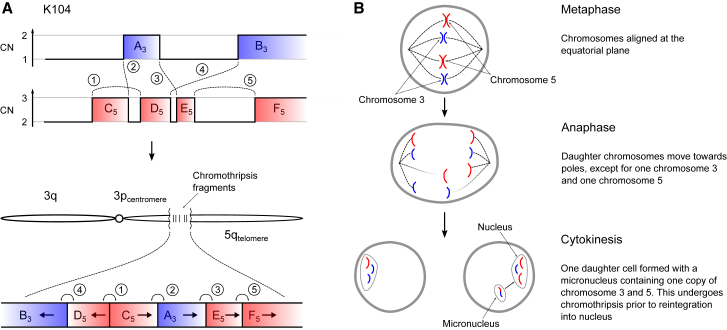
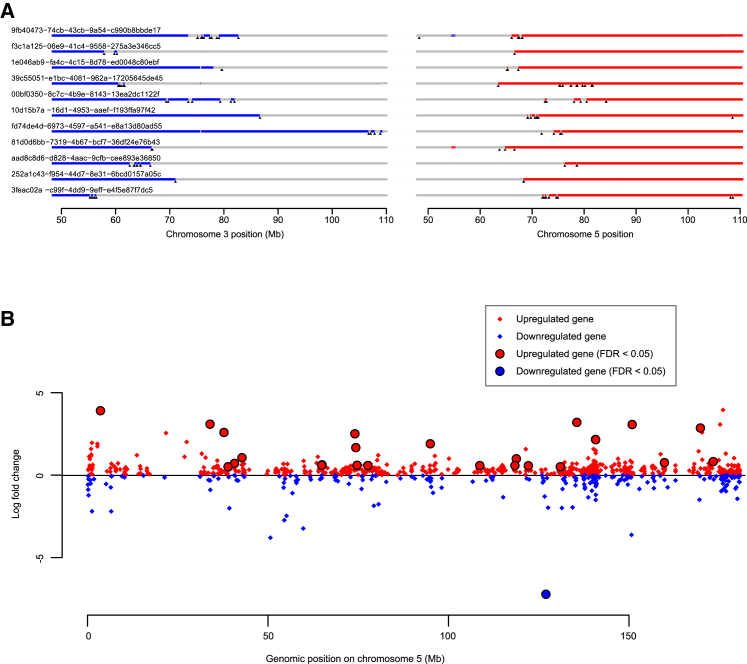
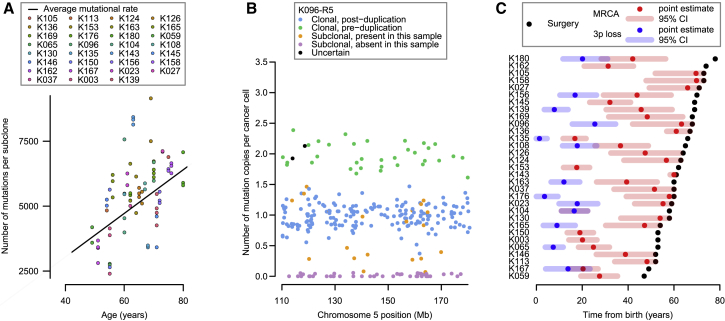
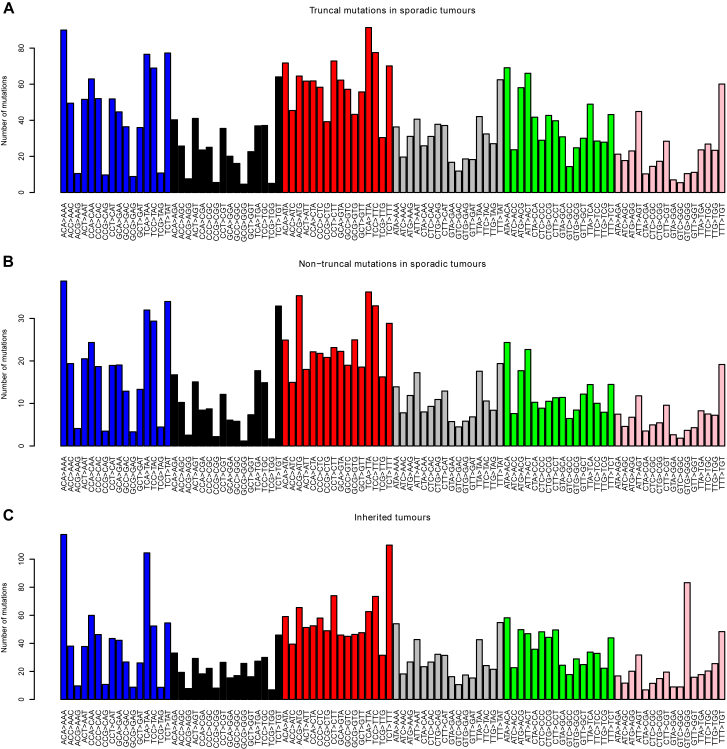
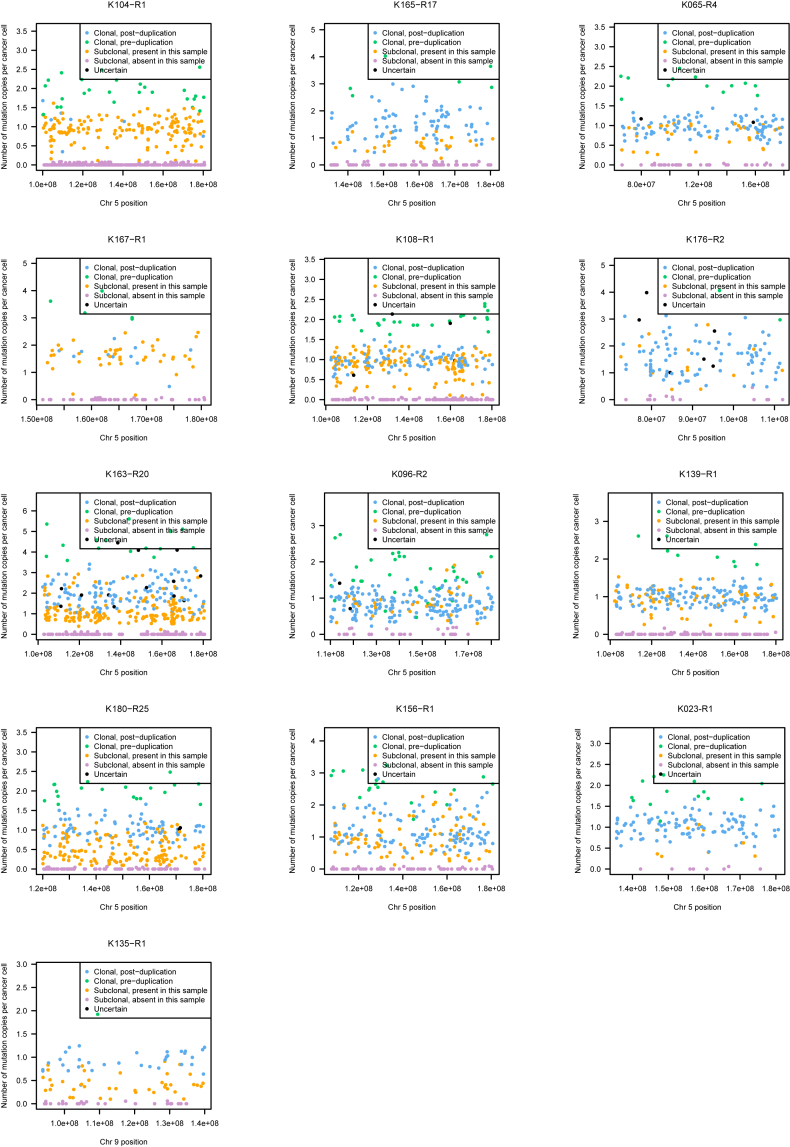
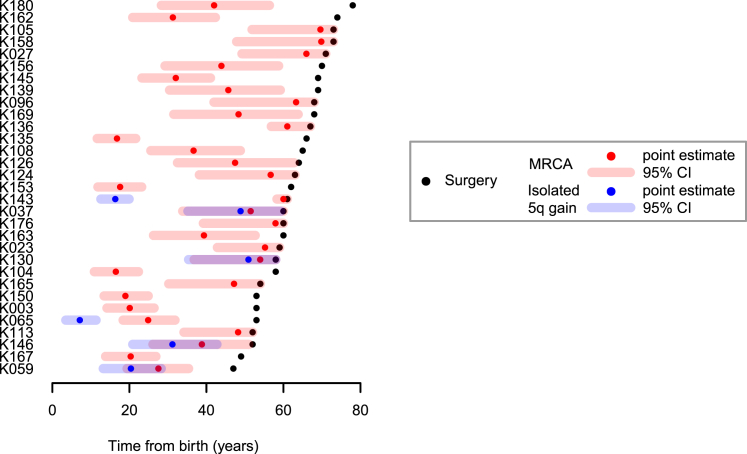
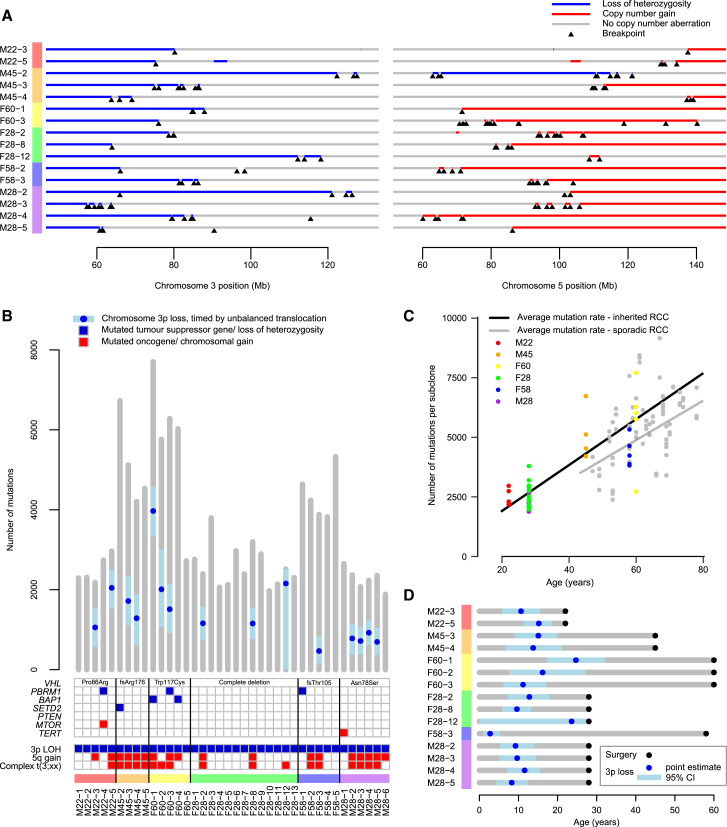
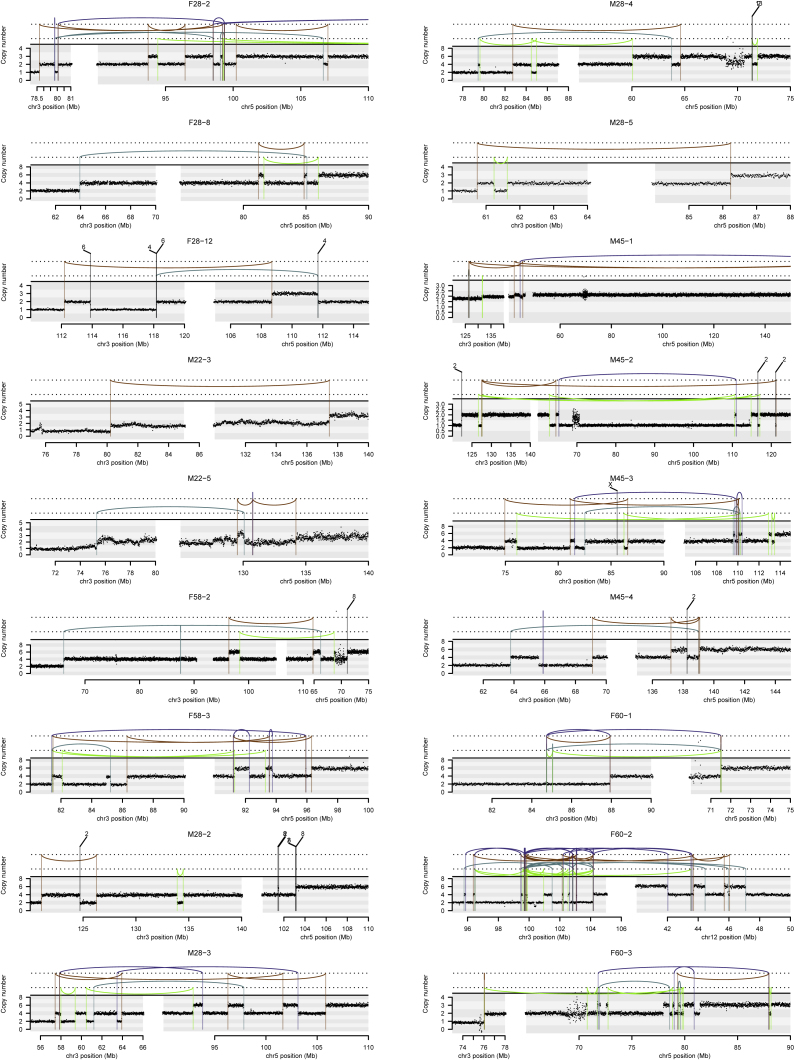
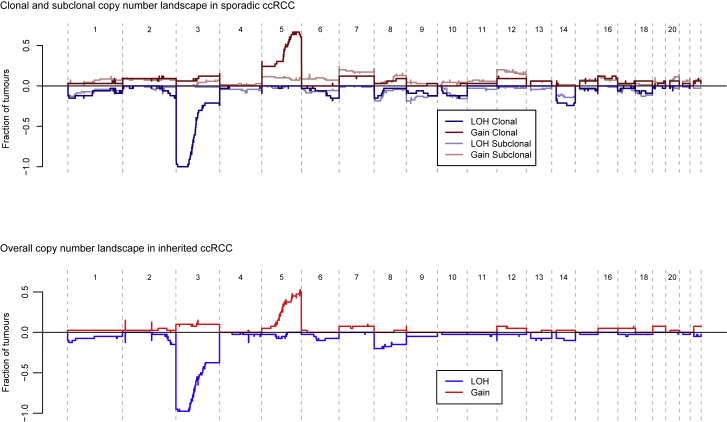
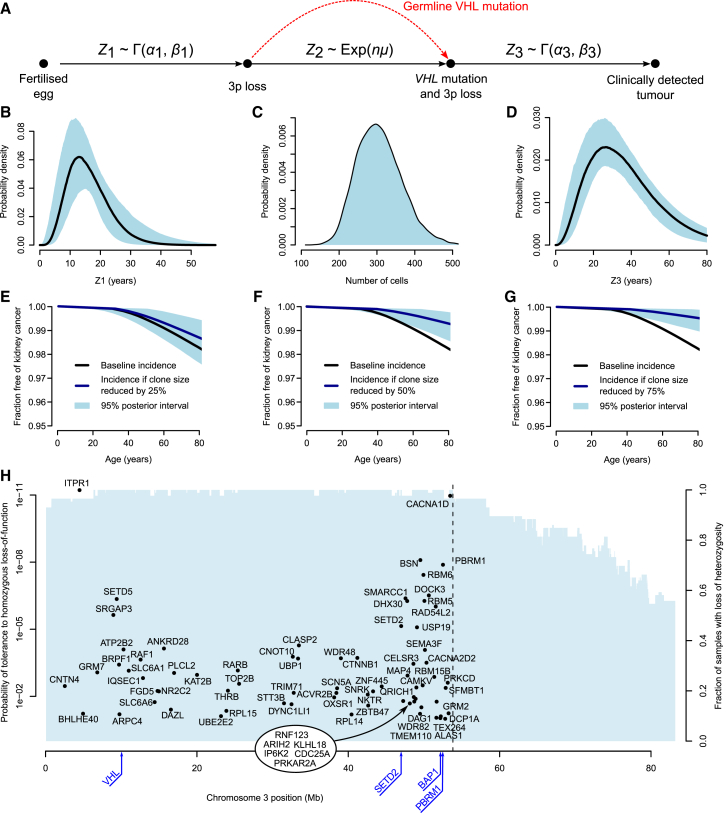
Clear cell renal cell carcinoma (ccRCC) is characterized by near-universal loss of the short arm of chromosome 3, deleting several tumor suppressor genes. We analyzed whole genomes from 95 biopsies across 33 patients with clear cell renal cell carcinoma. We find hotspots of point mutations in the 5' UTR of TERT, targeting a MYC-MAX-MAD1 repressor associated with telomere lengthening. The most common structural abnormality generates simultaneous 3p loss and 5q gain (36% patients), typically through chromothripsis. This event occurs in childhood or adolescence, generally as the initiating event that precedes emergence of the tumor's most recent common ancestor by years to decades. Similar genomic changes drive inherited ccRCC. Modeling differences in age incidence between inherited and sporadic cancers suggests that the number of cells with 3p loss capable of initiating sporadic tumors is no more than a few hundred. Early development of ccRCC follows well-defined evolutionary trajectories, offering opportunity for early intervention.
Keywords: cancer evolution; chromothripsis; clear cell renal cell carcinoma.
Crown Copyright © 2018. Published by Elsevier Inc. All rights reserved.
Figures

Comment in
-
Multi-regional Sequencing Elucidates the Evolution of Clear Cell Renal Cell Carcinoma.Cell. 2018 Apr 19;173(3):540-542. doi: 10.1016/j.cell.2018.03.077. Cell. 2018. PMID: 29677504
-
Tracing the steps of cancer evolution.Nat Rev Clin Oncol. 2018 Jul;15(7):401. doi: 10.1038/s41571-018-0033-z. Nat Rev Clin Oncol. 2018. PMID: 29700380 No abstract available.
-
Tracing clear cell renal carcinoma evolution.Nat Med. 2018 Jun;24(6):702. doi: 10.1038/s41591-018-0074-y. Nat Med. 2018. PMID: 29875457 No abstract available.
-
The origin, evolution and route to metastasis of clear cell RCC.Nat Rev Nephrol. 2018 Sep;14(9):538-540. doi: 10.1038/s41581-018-0031-5. Nat Rev Nephrol. 2018. PMID: 29875480 No abstract available.
References
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
-
- Ali H., Daser A., Dear P., Wood H., Rabbitts P., Rabbitts T. Nonreciprocal chromosomal translocations in renal cancer involve multiple DSBs and NHEJ associated with breakpoint inversion but not necessarily with transcription. Genes Chromosomes Cancer. 2013;52:402–409. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
