

Advanced oxidation protein products induce pre-osteoblast apoptosis through a nicotinamide adenine dinucleotide phosphate oxidase-dependent, mitogen-activated protein kinases-mediated intrinsic apoptosis pathway
- PMID: 29659123
- PMCID: PMC6052394
- DOI: 10.1111/acel.12764
Advanced oxidation protein products induce pre-osteoblast apoptosis through a nicotinamide adenine dinucleotide phosphate oxidase-dependent, mitogen-activated protein kinases-mediated intrinsic apoptosis pathway
Abstract
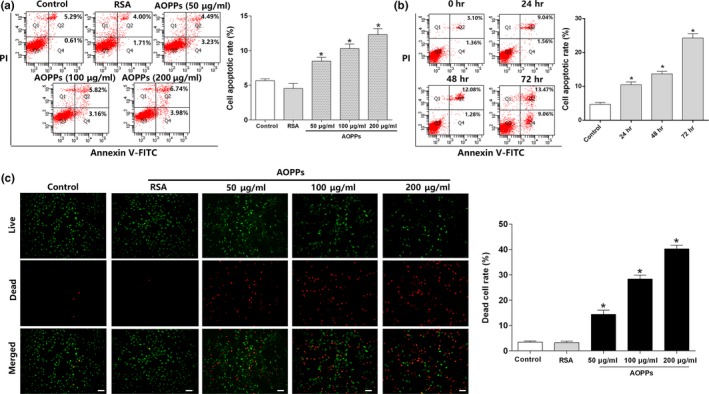
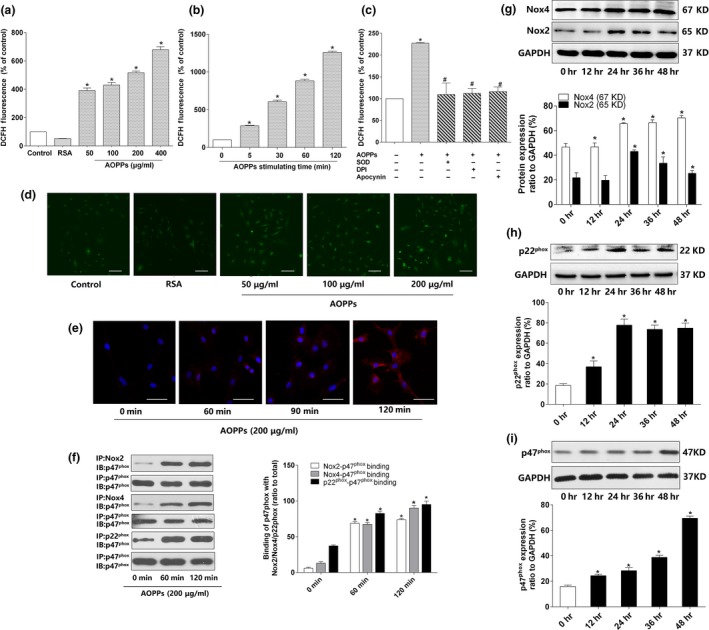
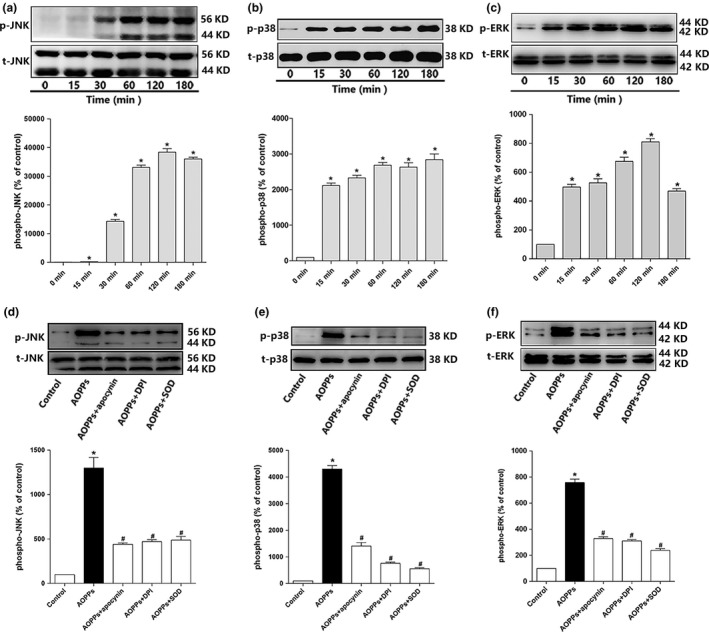
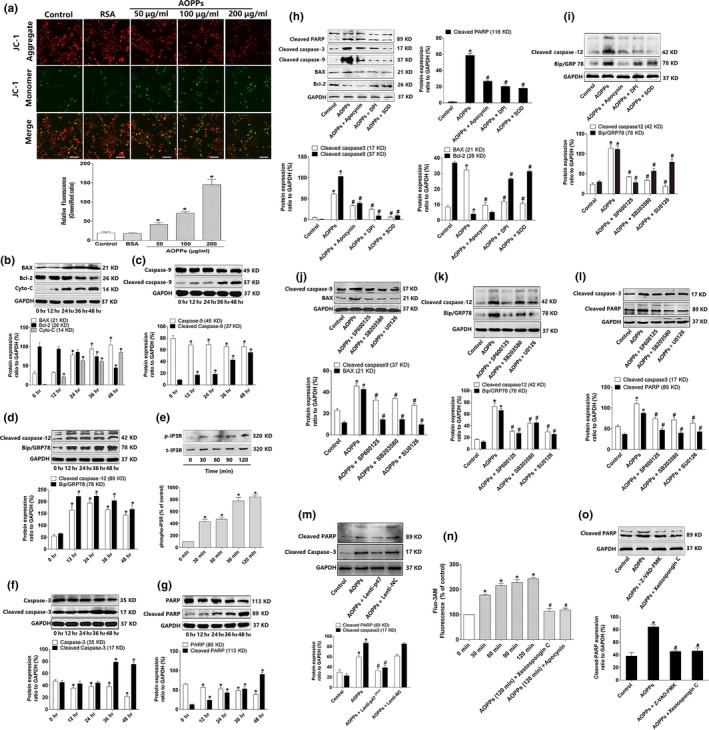
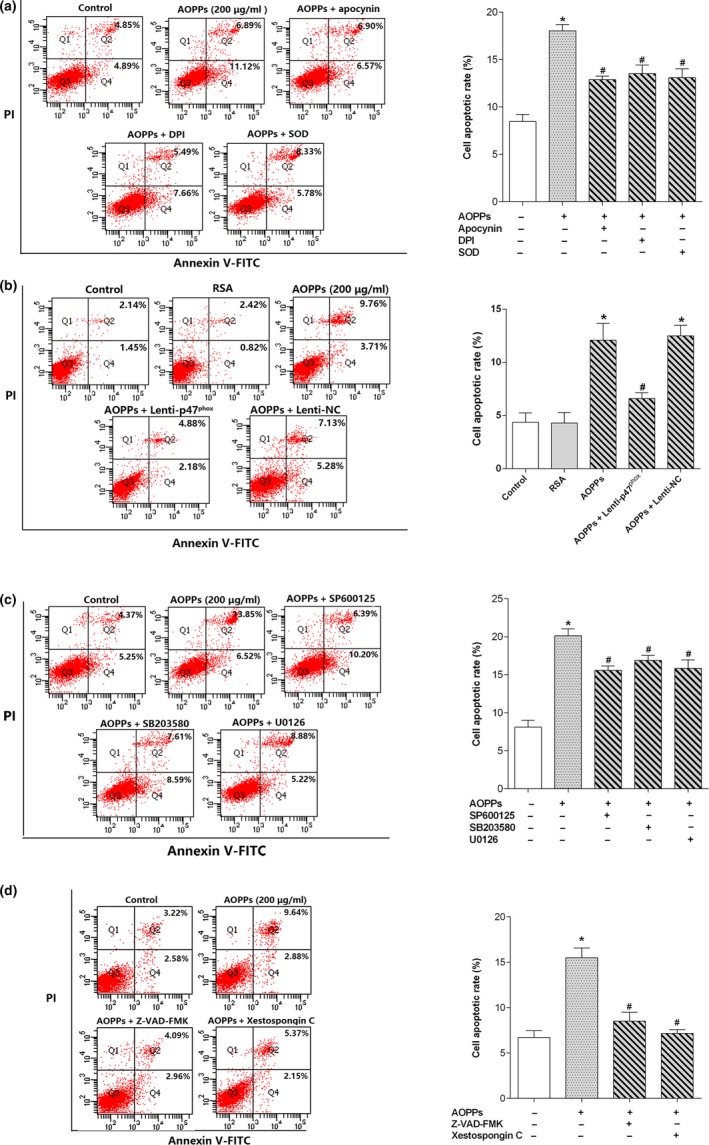
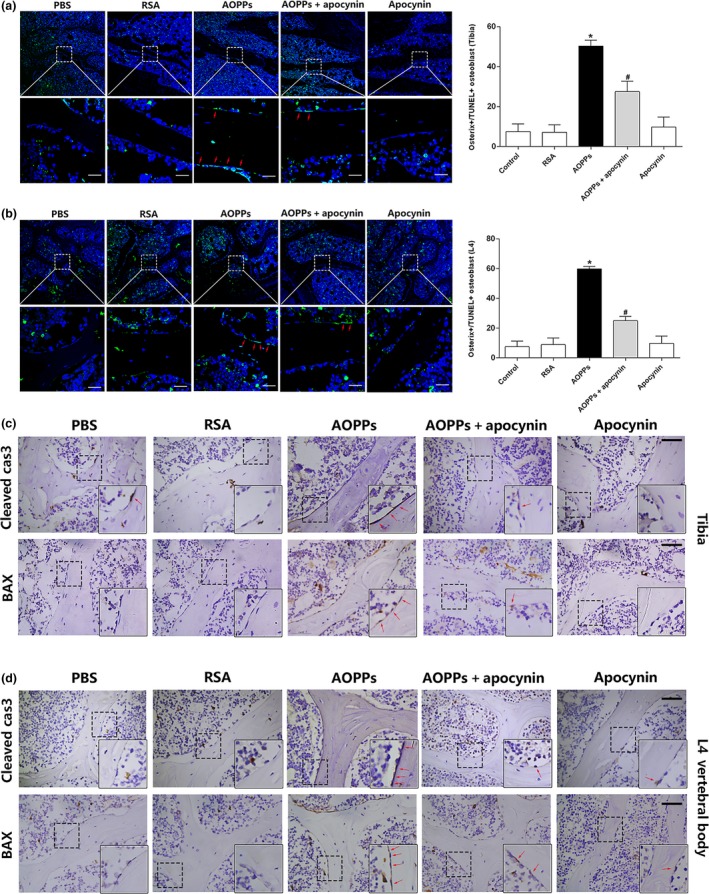
Osteoblast apoptosis contributes to age-related bone loss. Advanced oxidation protein products (AOPPs) are recognized as the markers of oxidative stress and potent inducers of apoptosis. We have demonstrated that AOPP accumulation was correlated with age-related bone loss. However, the effect of AOPPs on the osteoblast apoptosis still remains unknown. Exposure of osteoblastic MC3T3-E1 cells to AOPPs caused the excessive generation of reactive oxygen species (ROS) by activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. Increased ROS induced phosphorylation of mitogen-activated protein kinases (MAPKs), which subsequently triggered intrinsic apoptosis pathway by inducing mitochondrial dysfunction, endoplasmic reticulum stress, and Ca2+ overload and eventually leads to apoptosis. Chronic AOPP loading in aged Sprague-Dawley rats induced osteoblast apoptosis and activated NADPH oxidase signaling cascade, in combination with accelerated bone loss and deteriorated bone microstructure. Our study suggests that AOPPs induce osteoblast apoptosis by the NADPH oxidase-dependent, MAPK-mediated intrinsic apoptosis pathway.
Keywords: advanced oxidation protein products; apoptosis; osteoblast; osteoporosis; oxidative stress; reactive oxygen species.
© 2018 The Authors. Aging Cell published by the Anatomical Society and John Wiley & Sons Ltd.
Figures

References
-
- Chen, J. R. , Lazarenko, O. P. , Blackburn, M. L. , Mercer, K. E. , Badger, T. M. , & Ronis, M. J. (2015). p47phox‐Nox2‐dependent ROS signaling inhibits early bone development in mice but protects against skeletal aging. Journal of Biological Chemistry, 290, 14692–14704. 10.1074/jbc.m114.633461 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
