

Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm
- PMID: 29662057
- PMCID: PMC5902599
- DOI: 10.1038/s41467-018-03843-3
Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm
Abstract
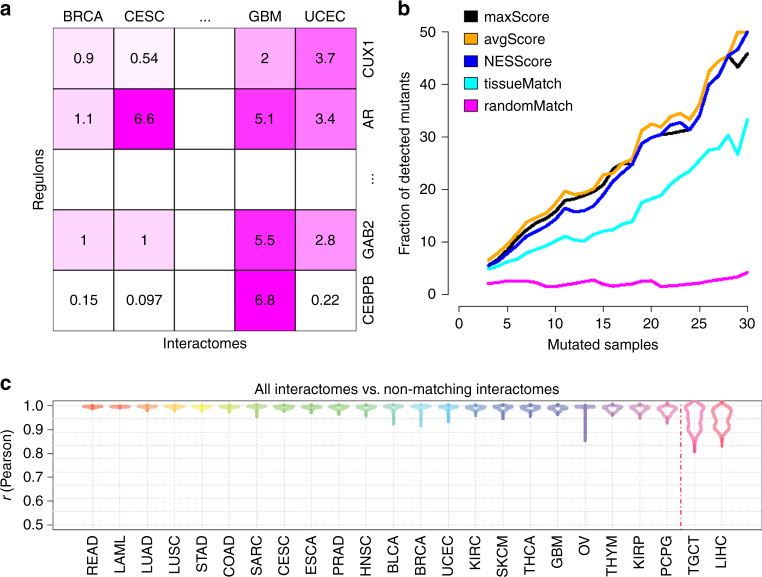
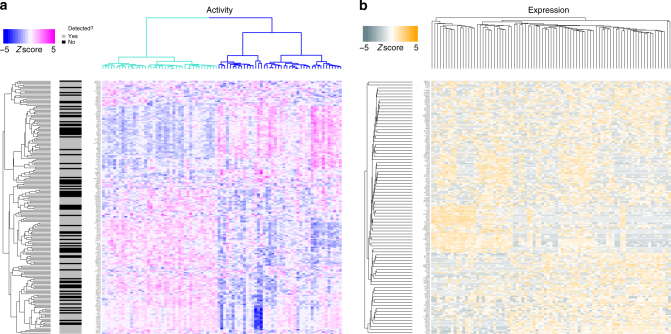
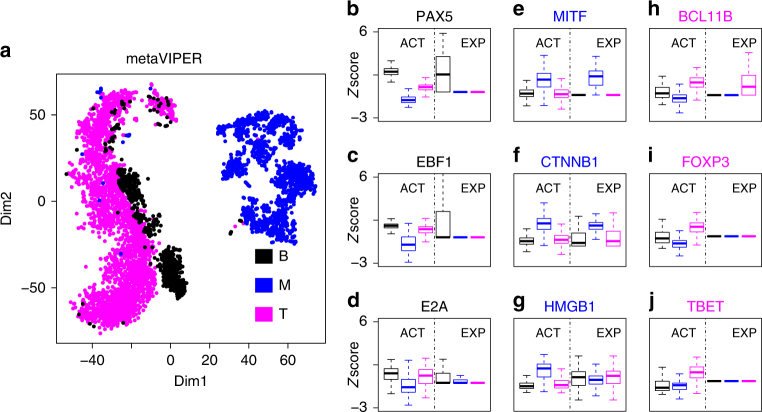
We and others have shown that transition and maintenance of biological states is controlled by master regulator proteins, which can be inferred by interrogating tissue-specific regulatory models (interactomes) with transcriptional signatures, using the VIPER algorithm. Yet, some tissues may lack molecular profiles necessary for interactome inference (orphan tissues), or, as for single cells isolated from heterogeneous samples, their tissue context may be undetermined. To address this problem, we introduce metaVIPER, an algorithm designed to assess protein activity in tissue-independent fashion by integrative analysis of multiple, non-tissue-matched interactomes. This assumes that transcriptional targets of each protein will be recapitulated by one or more available interactomes. We confirm the algorithm's value in assessing protein dysregulation induced by somatic mutations, as well as in assessing protein activity in orphan tissues and, most critically, in single cells, thus allowing transformation of noisy and potentially biased RNA-Seq signatures into reproducible protein-activity signatures.
Conflict of interest statement
M.J.A. is chief scientific officer of DarwinHealth Inc. A.C. is founder and equity holder of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc. The remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
