

Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets
- PMID: 29662167
- PMCID: PMC6372064
- DOI: 10.1038/s41588-018-0086-z
Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets
Abstract
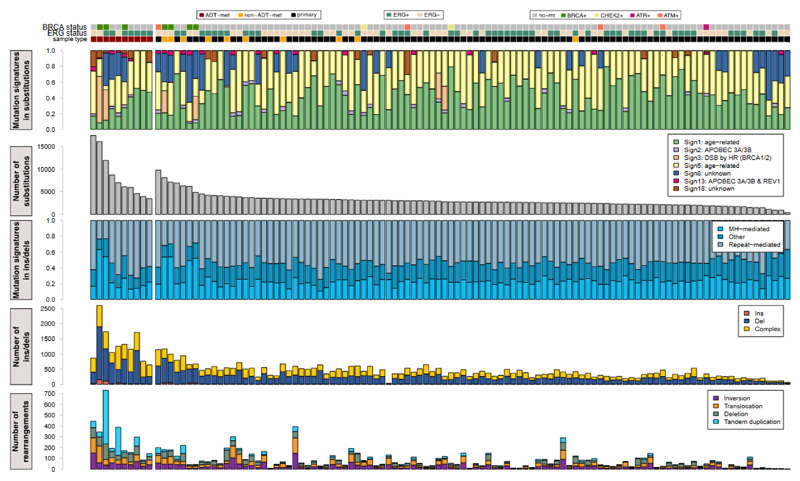
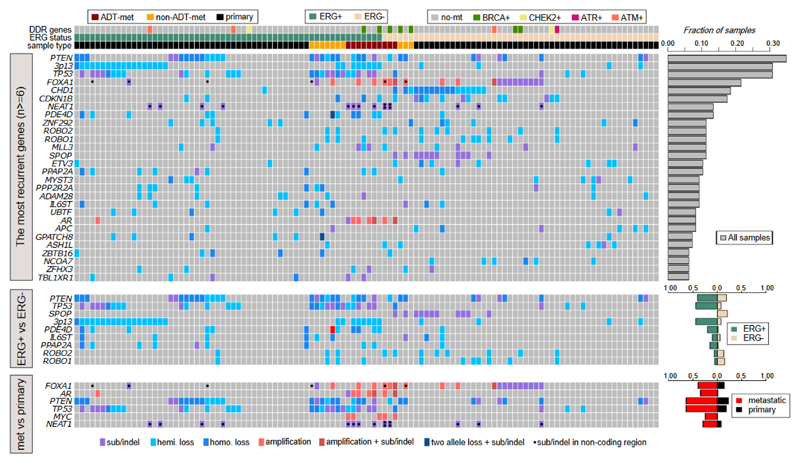
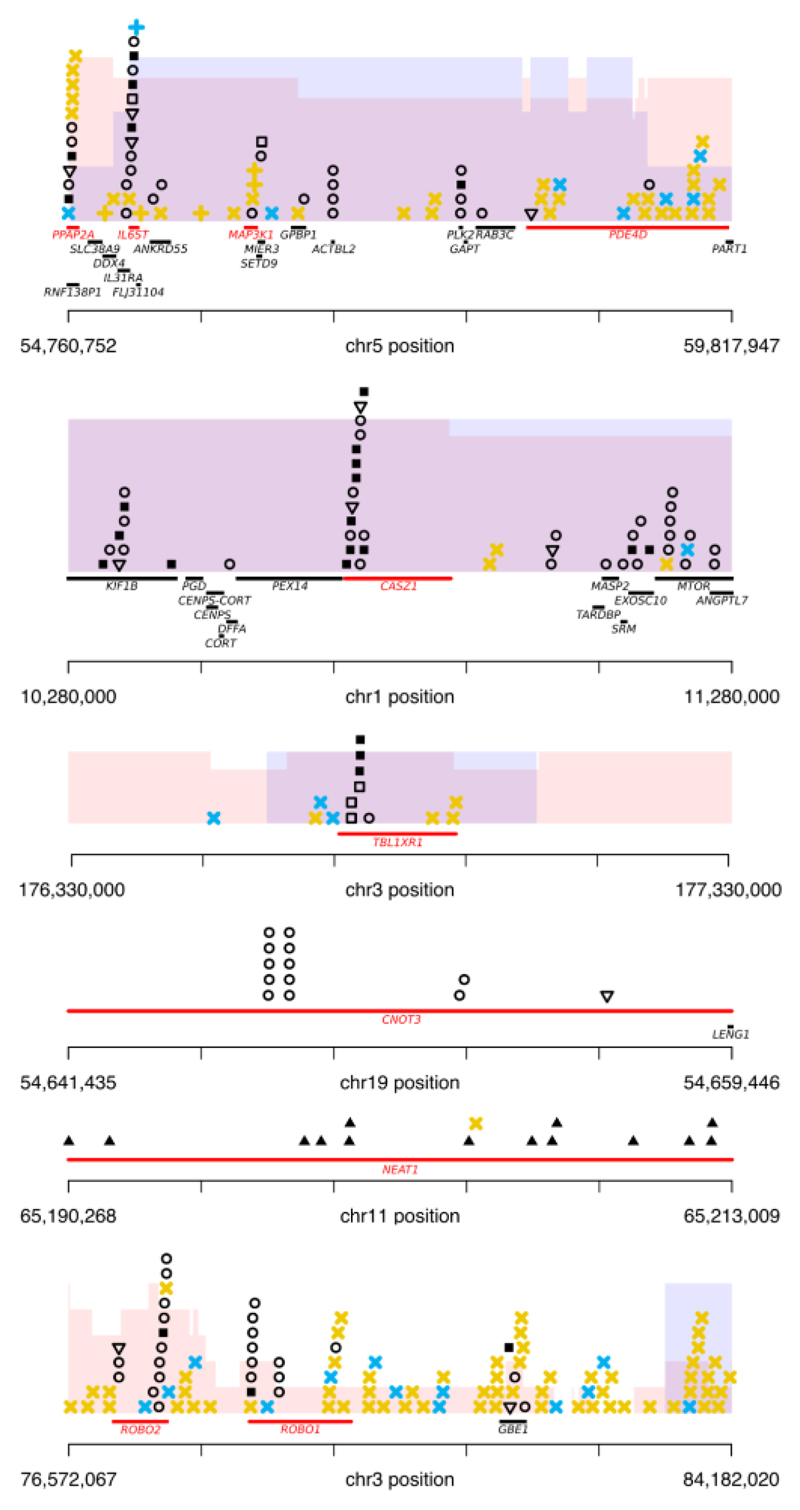
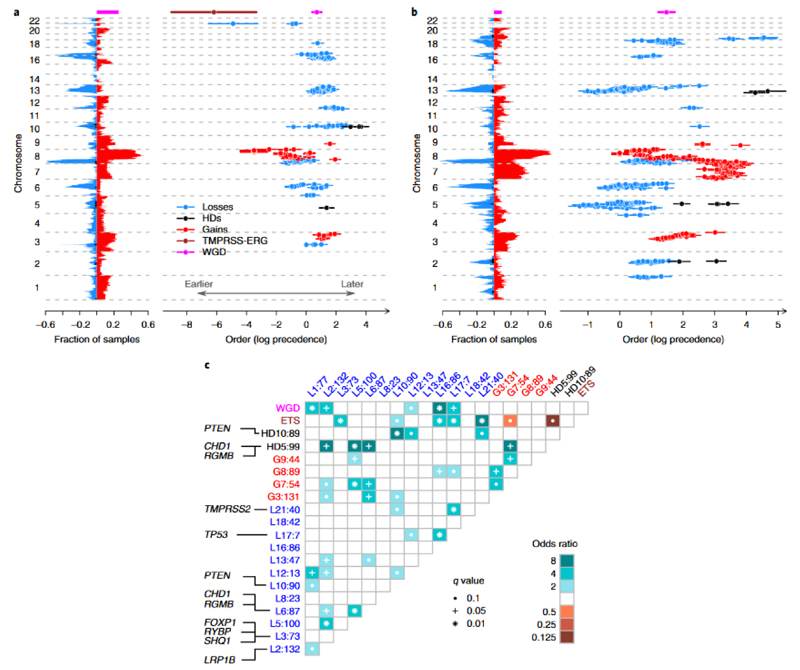
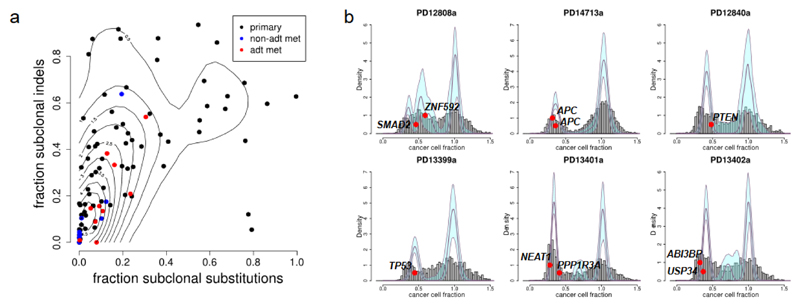
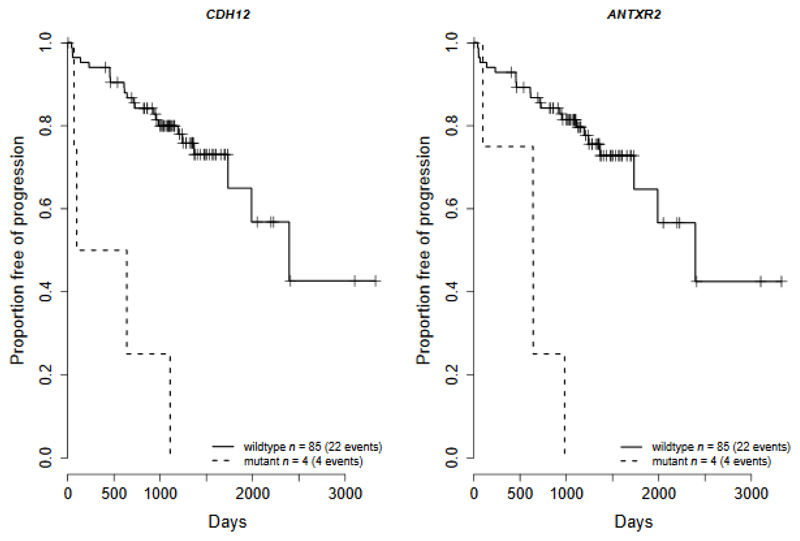
Prostate cancer represents a substantial clinical challenge because it is difficult to predict outcome and advanced disease is often fatal. We sequenced the whole genomes of 112 primary and metastatic prostate cancer samples. From joint analysis of these cancers with those from previous studies (930 cancers in total), we found evidence for 22 previously unidentified putative driver genes harboring coding mutations, as well as evidence for NEAT1 and FOXA1 acting as drivers through noncoding mutations. Through the temporal dissection of aberrations, we identified driver mutations specifically associated with steps in the progression of prostate cancer, establishing, for example, loss of CHD1 and BRCA2 as early events in cancer development of ETS fusion-negative cancers. Computational chemogenomic (canSAR) analysis of prostate cancer mutations identified 11 targets of approved drugs, 7 targets of investigational drugs, and 62 targets of compounds that may be active and should be considered candidates for future clinical trials.
Conflict of interest statement
There are no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- 17528/CRUK_/Cancer Research UK/United Kingdom
- 22897/CRUK_/Cancer Research UK/United Kingdom
- A17528/CRUK_/Cancer Research UK/United Kingdom
- A7990/CRUK_/Cancer Research UK/United Kingdom
- 26718/CRUK_/Cancer Research UK/United Kingdom
- A14835/CRUK_/Cancer Research UK/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- 11566/CRUK_/Cancer Research UK/United Kingdom
- 22744/CRUK_/Cancer Research UK/United Kingdom
- A22530/CRUK_/Cancer Research UK/United Kingdom
- 22530/CRUK_/Cancer Research UK/United Kingdom
- 20406/CRUK_/Cancer Research UK/United Kingdom
- 21777/CRUK_/Cancer Research UK/United Kingdom
- A6743/CRUK_/Cancer Research UK/United Kingdom
- A23187/CRUK_/Cancer Research UK/United Kingdom
- A11566/CRUK_/Cancer Research UK/United Kingdom
- P30 CA008748/CA/NCI NIH HHS/United States
- 23187/CRUK_/Cancer Research UK/United Kingdom
- 15007/CRUK_/Cancer Research UK/United Kingdom
- 14835/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
