

Unifying cancer and normal RNA sequencing data from different sources
- PMID: 29664468
- PMCID: PMC5903355
- DOI: 10.1038/sdata.2018.61
Unifying cancer and normal RNA sequencing data from different sources
Abstract
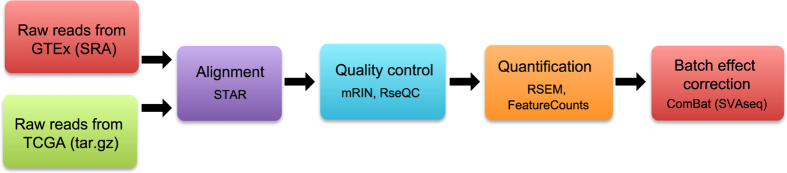
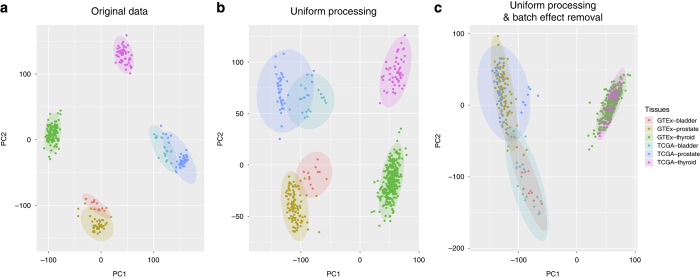
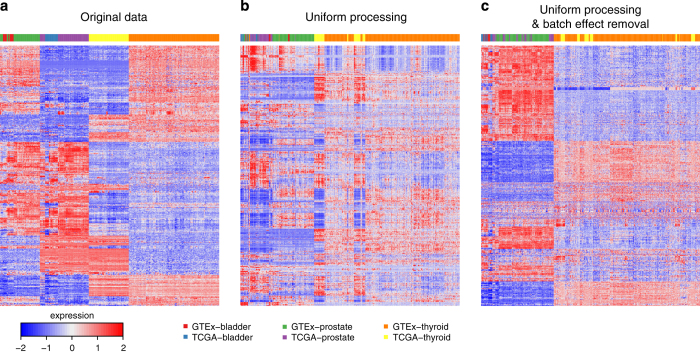
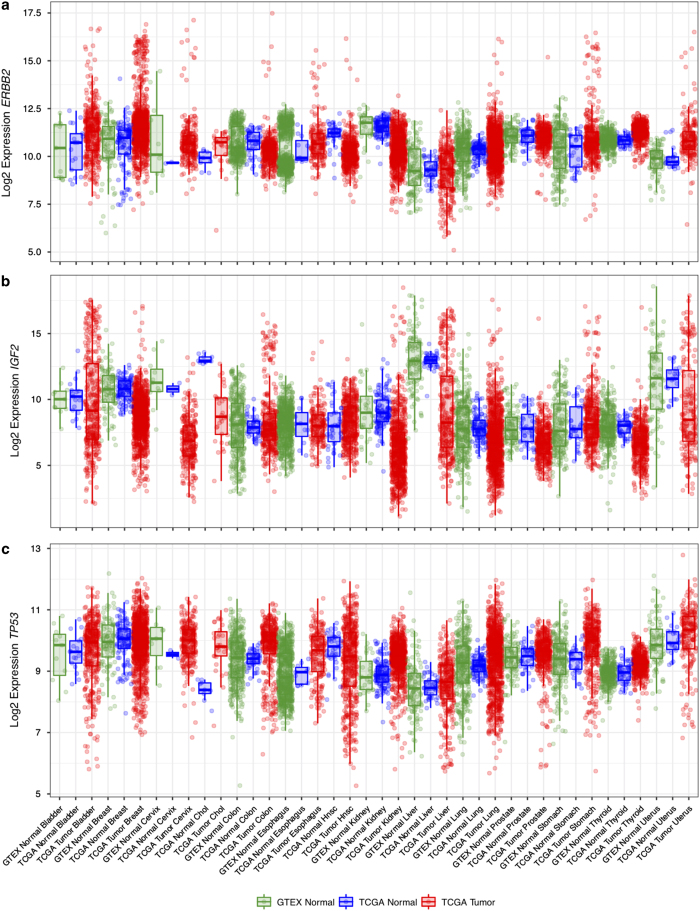
Driven by the recent advances of next generation sequencing (NGS) technologies and an urgent need to decode complex human diseases, a multitude of large-scale studies were conducted recently that have resulted in an unprecedented volume of whole transcriptome sequencing (RNA-seq) data, such as the Genotype Tissue Expression project (GTEx) and The Cancer Genome Atlas (TCGA). While these data offer new opportunities to identify the mechanisms underlying disease, the comparison of data from different sources remains challenging, due to differences in sample and data processing. Here, we developed a pipeline that processes and unifies RNA-seq data from different studies, which includes uniform realignment, gene expression quantification, and batch effect removal. We find that uniform alignment and quantification is not sufficient when combining RNA-seq data from different sources and that the removal of other batch effects is essential to facilitate data comparison. We have processed data from GTEx and TCGA and successfully corrected for study-specific biases, enabling comparative analysis between TCGA and GTEx. The normalized datasets are available for download on figshare.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Data Citations
-
- Wang Q., Gao J., Nikolaus S. 2017. Figshare. https://doi.org/10.6084/m9.figshare.5330539 - DOI
-
- Wang Q., Gao J., Nikolaus S. 2017. Figshare. https://doi.org/10.6084/m9.figshare.5330575 - DOI
-
- Wang Q., Gao J., Nikolaus S. 2017. Figshare. https://doi.org/10.6084/m9.figshare.5330593 - DOI
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
