

Emerging therapeutic targets for neurofibromatosis type 1
- PMID: 29667529
- PMCID: PMC7017752
- DOI: 10.1080/14728222.2018.1465931
Emerging therapeutic targets for neurofibromatosis type 1
Abstract
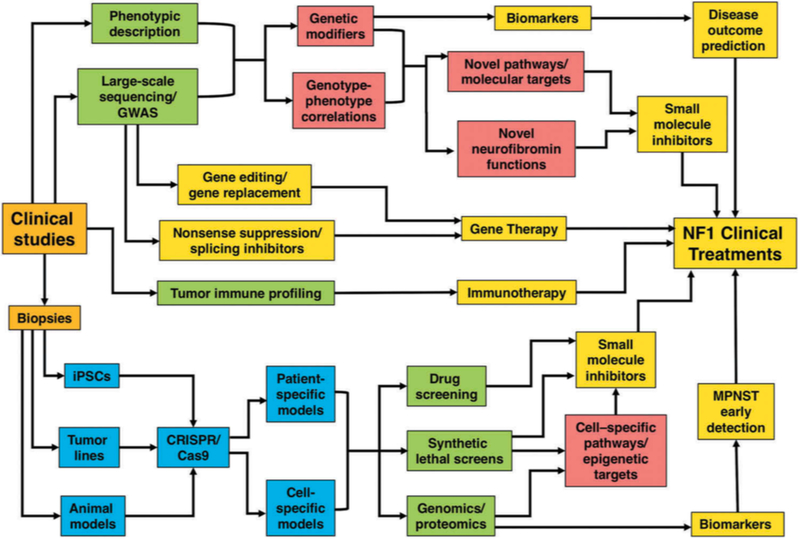
Neurofibromatosis type 1 (NF1) is an autosomal dominantly inherited tumor predisposition syndrome with an incidence of one in 3000-4000 individuals with no currently effective therapies. The NF1 gene encodes neurofibromin, which functions as a negative regulator of RAS. NF1 is a chronic multisystem disorder affecting many different tissues. Due to cell-specific complexities of RAS signaling, therapeutic approaches for NF1 will likely have to focus on a particular tissue and manifestation of the disease. Areas covered: We discuss the multisystem nature of NF1 and the signaling pathways affected due to neurofibromin deficiency. We explore the cell-/tissue-specific molecular and cellular consequences of aberrant RAS signaling in NF1 and speculate on their potential as therapeutic targets for the disease. We discuss recent genomic, transcriptomic, and proteomic studies combined with molecular, cellular, and biochemical analyses which have identified several targets for specific NF1 manifestations. We also consider the possibility of patient-specific gene therapy approaches for NF1. Expert opinion: The emergence of NF1 genotype-phenotype correlations, characterization of cell-specific signaling pathways affected in NF1, identification of novel biomarkers, and the development of sophisticated animal models accurately reflecting human pathology will continue to provide opportunities to develop therapeutic approaches to combat this multisystem disorder.
Keywords: Neurofibromas; RAS-MEK-ERK signaling; RASopathies; neurofibromatosis type 1; peripheral nerve sheath tumors.
Figures
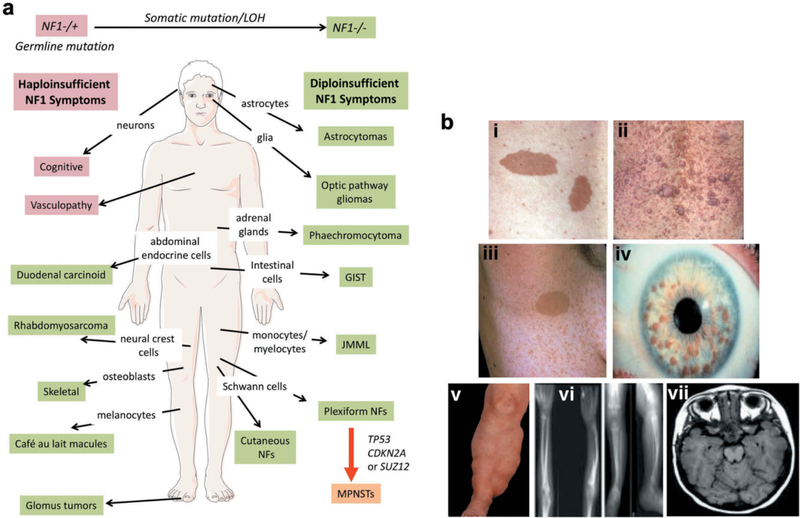
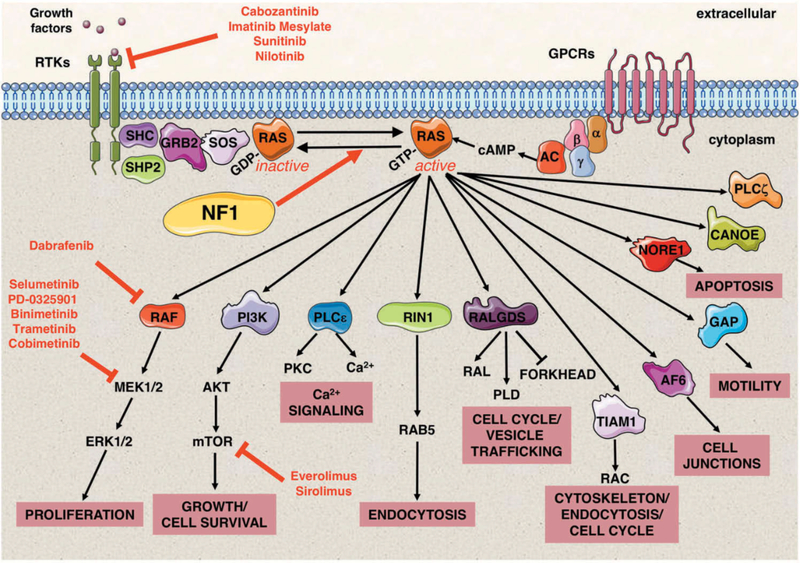
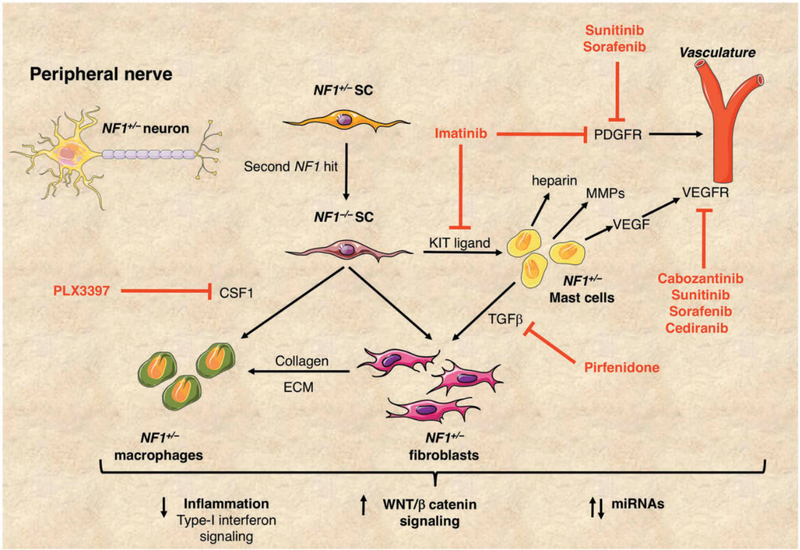
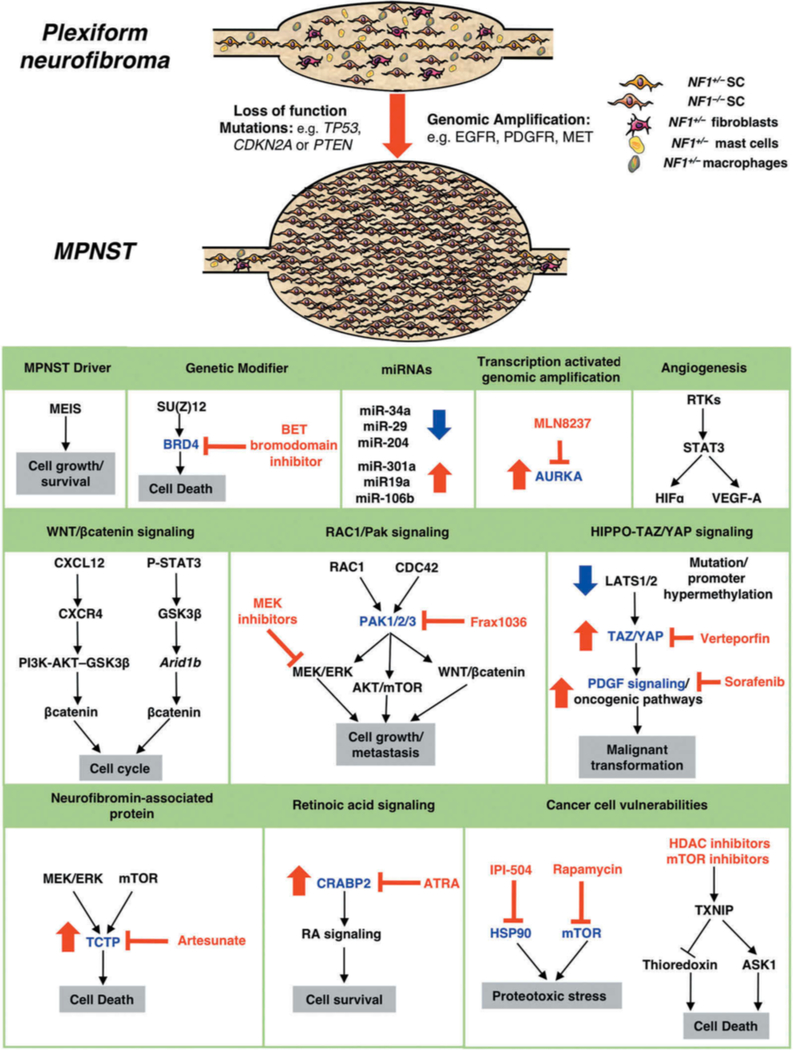
References
-
- Upadhyaya M, Cooper DN. “Molecular and Cellular Biology of Neurofibromatosis Type 1”. 2012:Eds: Upadhyaya M and Cooper DN Springer Verlag.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous