

Identification of residue pairing in interacting β-strands from a predicted residue contact map
- PMID: 29673311
- PMCID: PMC5907701
- DOI: 10.1186/s12859-018-2150-1
Identification of residue pairing in interacting β-strands from a predicted residue contact map
Abstract
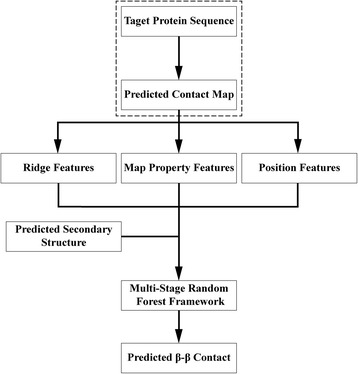
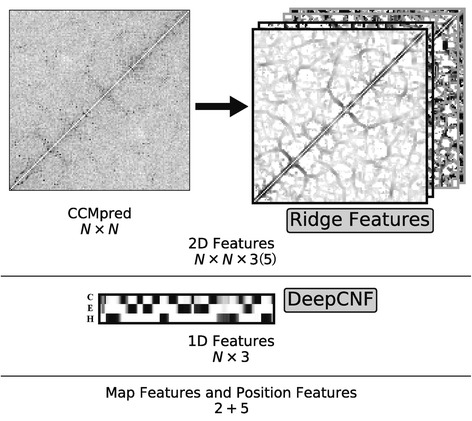
Background: Despite the rapid progress of protein residue contact prediction, predicted residue contact maps frequently contain many errors. However, information of residue pairing in β strands could be extracted from a noisy contact map, due to the presence of characteristic contact patterns in β-β interactions. This information may benefit the tertiary structure prediction of mainly β proteins. In this work, we propose a novel ridge-detection-based β-β contact predictor to identify residue pairing in β strands from any predicted residue contact map.
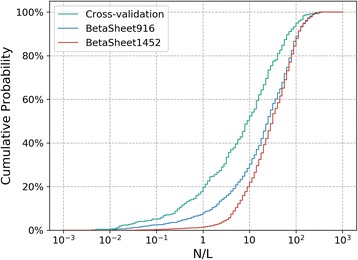
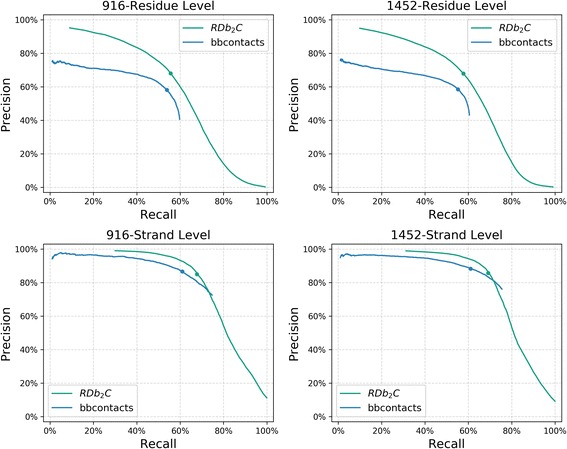
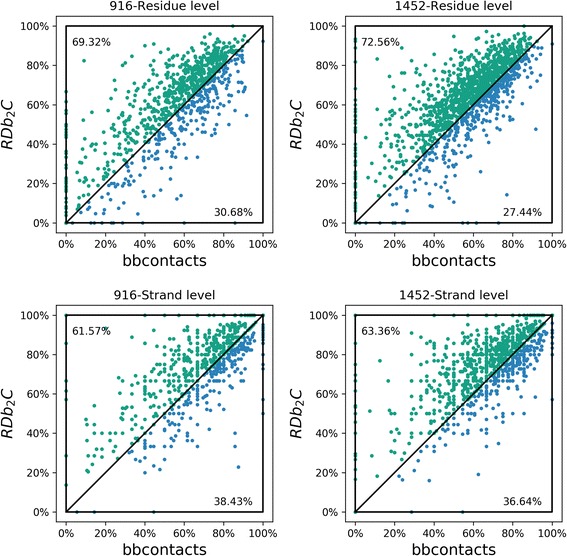
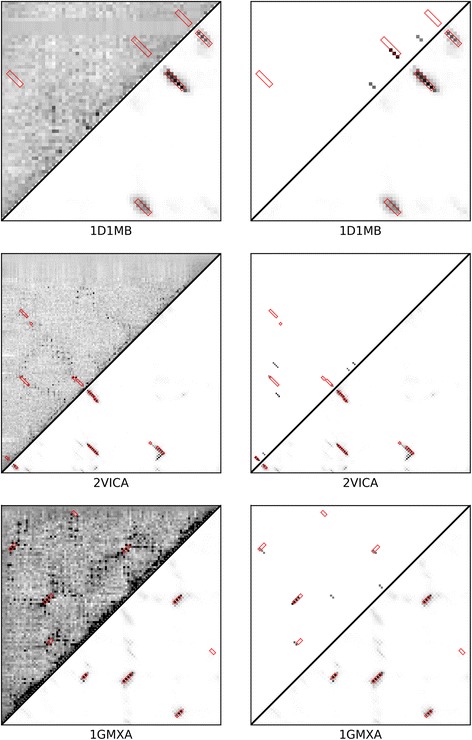
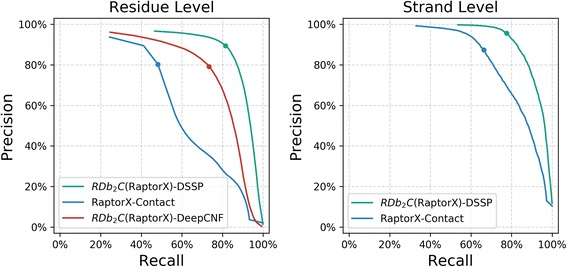
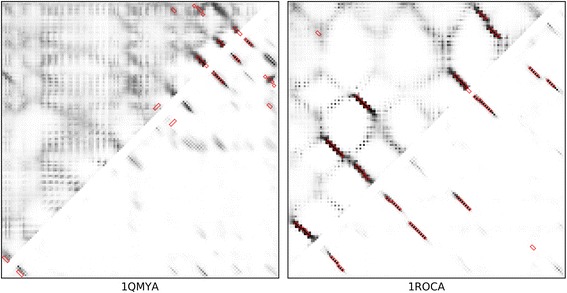
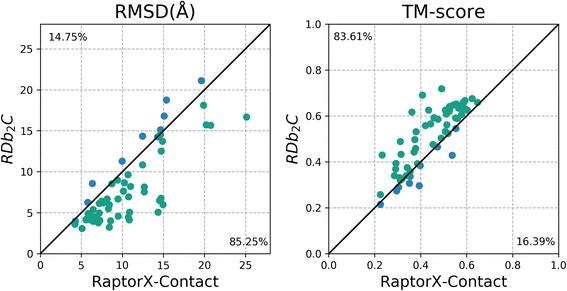
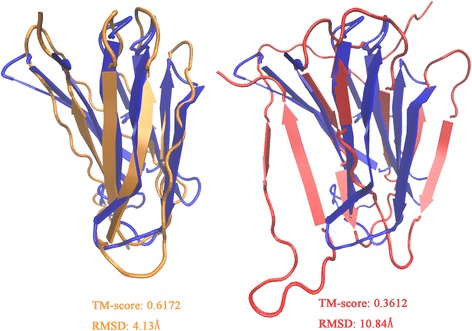
Results: Our algorithm RDb2C adopts ridge detection, a well-developed technique in computer image processing, to capture consecutive residue contacts, and then utilizes a novel multi-stage random forest framework to integrate the ridge information and additional features for prediction. Starting from the predicted contact map of CCMpred, RDb2C remarkably outperforms all state-of-the-art methods on two conventional test sets of β proteins (BetaSheet916 and BetaSheet1452), and achieves F1-scores of ~ 62% and ~ 76% at the residue level and strand level, respectively. Taking the prediction of the more advanced RaptorX-Contact as input, RDb2C achieves impressively higher performance, with F1-scores reaching ~ 76% and ~ 86% at the residue level and strand level, respectively. In a test of structural modeling using the top 1 L predicted contacts as constraints, for 61 mainly β proteins, the average TM-score achieves 0.442 when using the raw RaptorX-Contact prediction, but increases to 0.506 when using the improved prediction by RDb2C.
Conclusion: Our method can significantly improve the prediction of β-β contacts from any predicted residue contact maps. Prediction results of our algorithm could be directly applied to effectively facilitate the practical structure prediction of mainly β proteins.
Availability: All source data and codes are available at http://166.111.152.91/Downloads.html or the GitHub address of https://github.com/wzmao/RDb2C .
Keywords: Contact map; Protein structure prediction; Random forest; Residue contact prediction; Ridge detection; β-β pairing.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
