

A site specific model and analysis of the neutral somatic mutation rate in whole-genome cancer data
- PMID: 29673314
- PMCID: PMC5909259
- DOI: 10.1186/s12859-018-2141-2
A site specific model and analysis of the neutral somatic mutation rate in whole-genome cancer data
Abstract
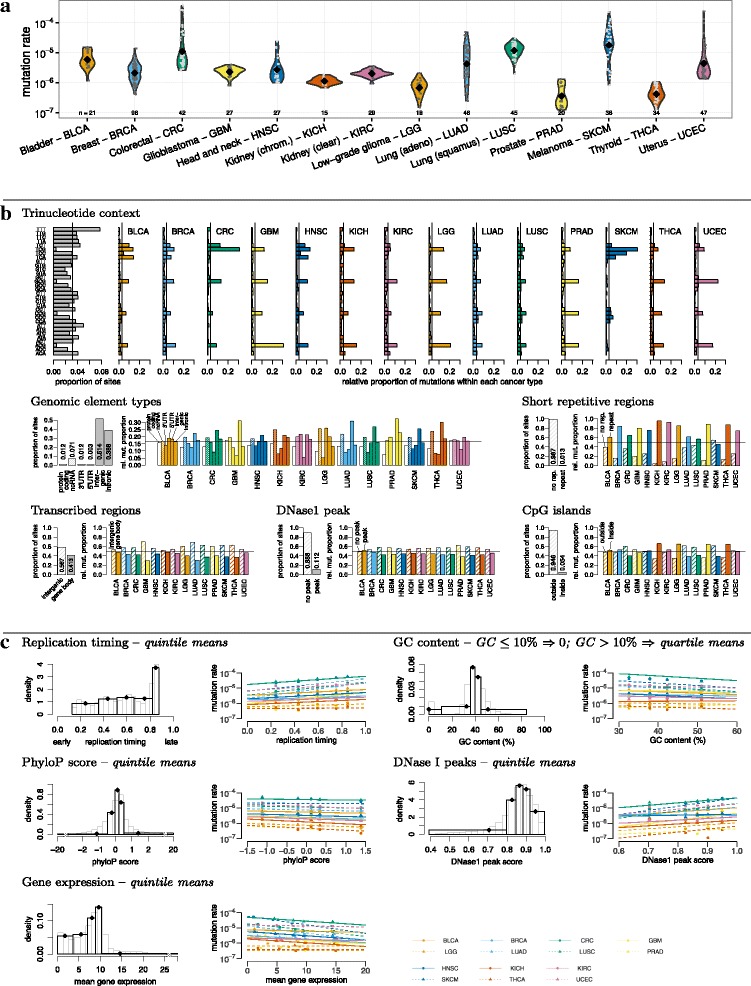
Background: Detailed modelling of the neutral mutational process in cancer cells is crucial for identifying driver mutations and understanding the mutational mechanisms that act during cancer development. The neutral mutational process is very complex: whole-genome analyses have revealed that the mutation rate differs between cancer types, between patients and along the genome depending on the genetic and epigenetic context. Therefore, methods that predict the number of different types of mutations in regions or specific genomic elements must consider local genomic explanatory variables. A major drawback of most methods is the need to average the explanatory variables across the entire region or genomic element. This procedure is particularly problematic if the explanatory variable varies dramatically in the element under consideration.
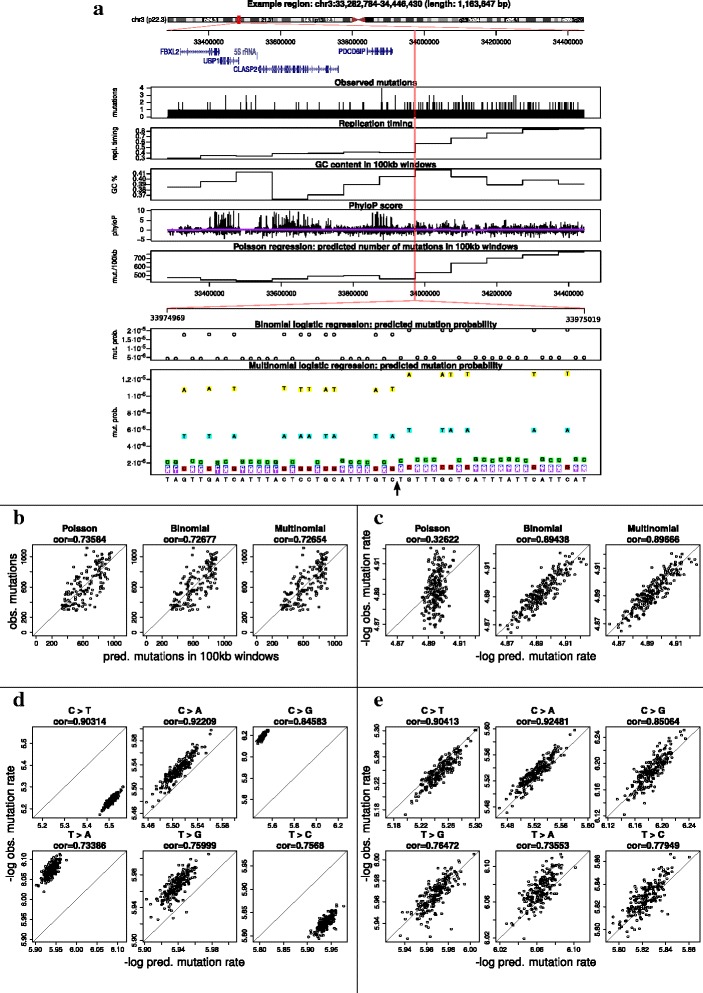
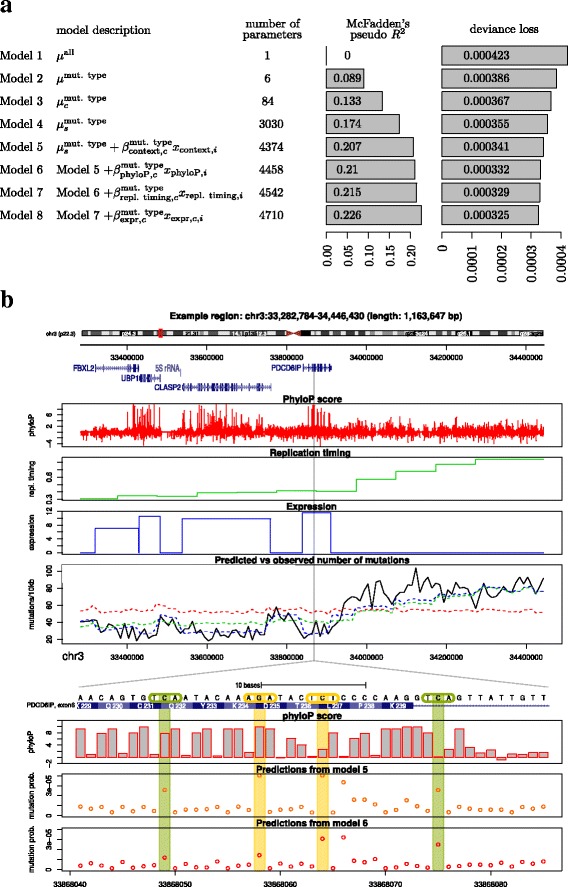
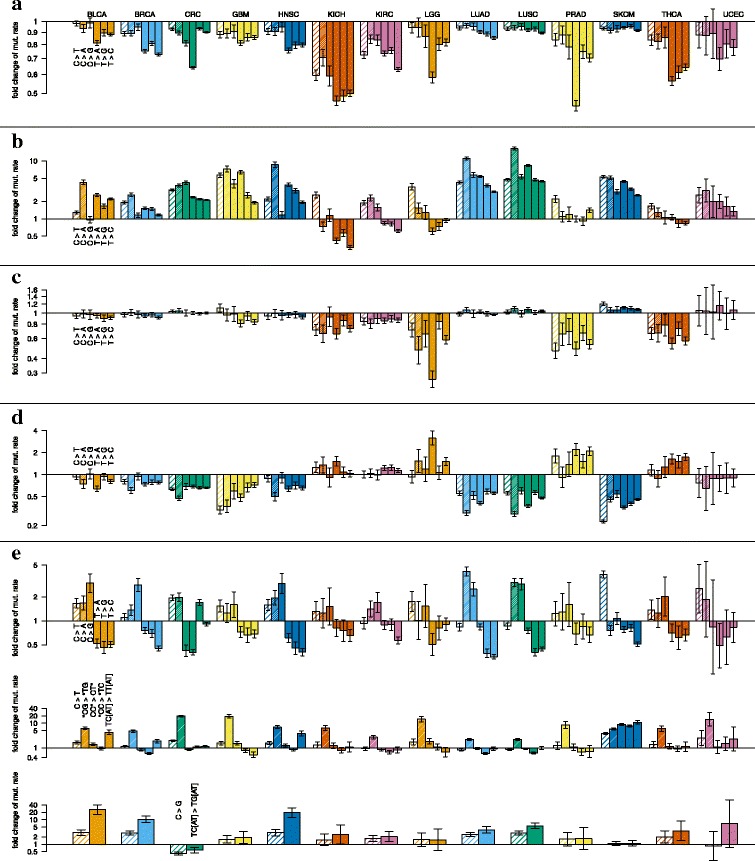
Results: To take into account the fine scale of the explanatory variables, we model the probabilities of different types of mutations for each position in the genome by multinomial logistic regression. We analyse 505 cancer genomes from 14 different cancer types and compare the performance in predicting mutation rate for both regional based models and site-specific models. We show that for 1000 randomly selected genomic positions, the site-specific model predicts the mutation rate much better than regional based models. We use a forward selection procedure to identify the most important explanatory variables. The procedure identifies site-specific conservation (phyloP), replication timing, and expression level as the best predictors for the mutation rate. Finally, our model confirms and quantifies certain well-known mutational signatures.
Conclusion: We find that our site-specific multinomial regression model outperforms the regional based models. The possibility of including genomic variables on different scales and patient specific variables makes it a versatile framework for studying different mutational mechanisms. Our model can serve as the neutral null model for the mutational process; regions that deviate from the null model are candidates for elements that drive cancer development.
Keywords: Multinomial logistic regression; Site-specific model; Somatic cancer mutations.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
