

Systematic analysis of complex genetic interactions
- PMID: 29674565
- PMCID: PMC6215713
- DOI: 10.1126/science.aao1729
Systematic analysis of complex genetic interactions
Abstract
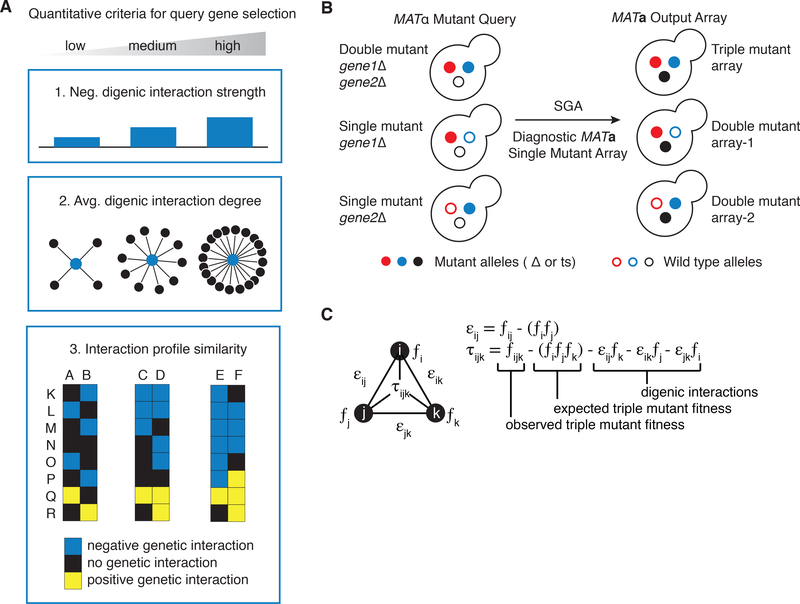
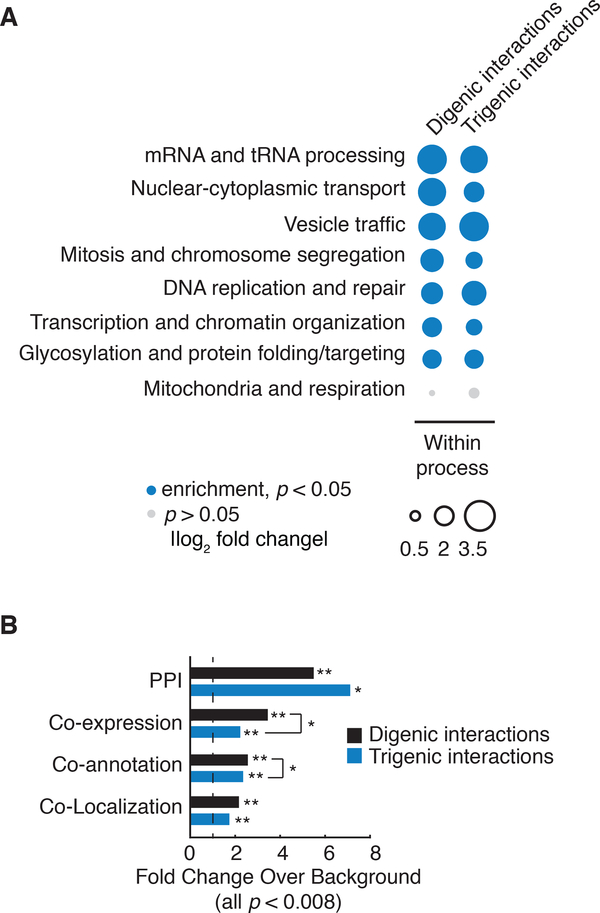
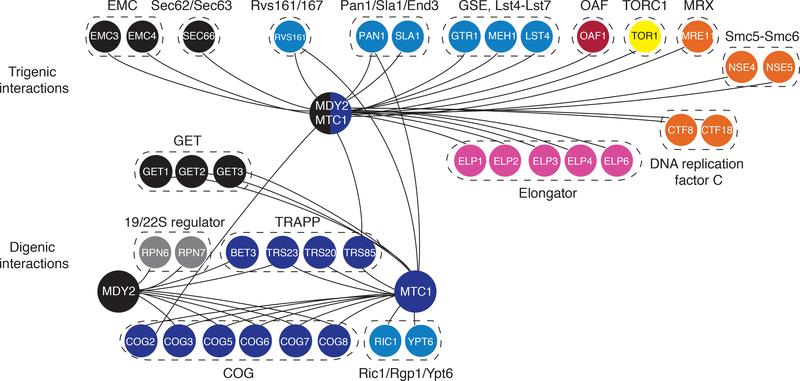
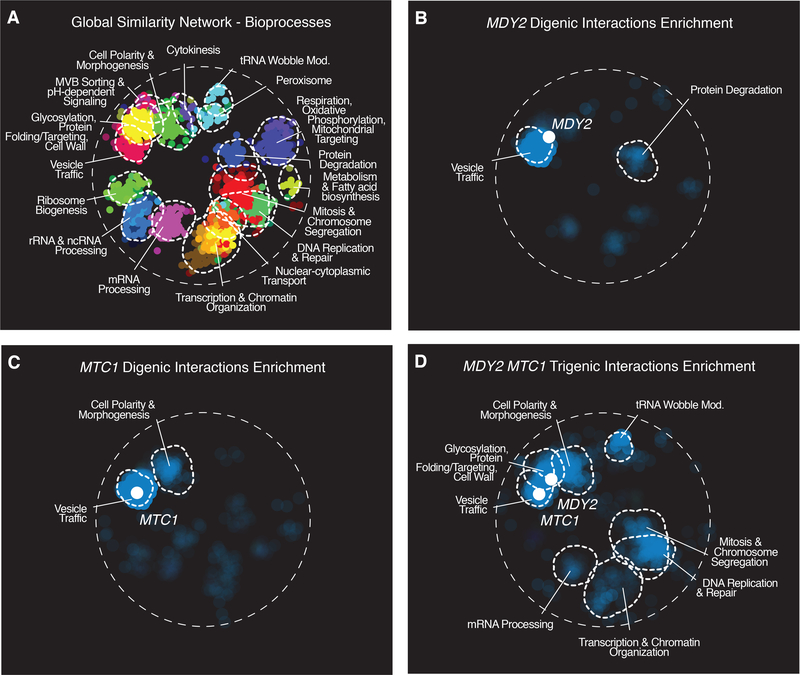
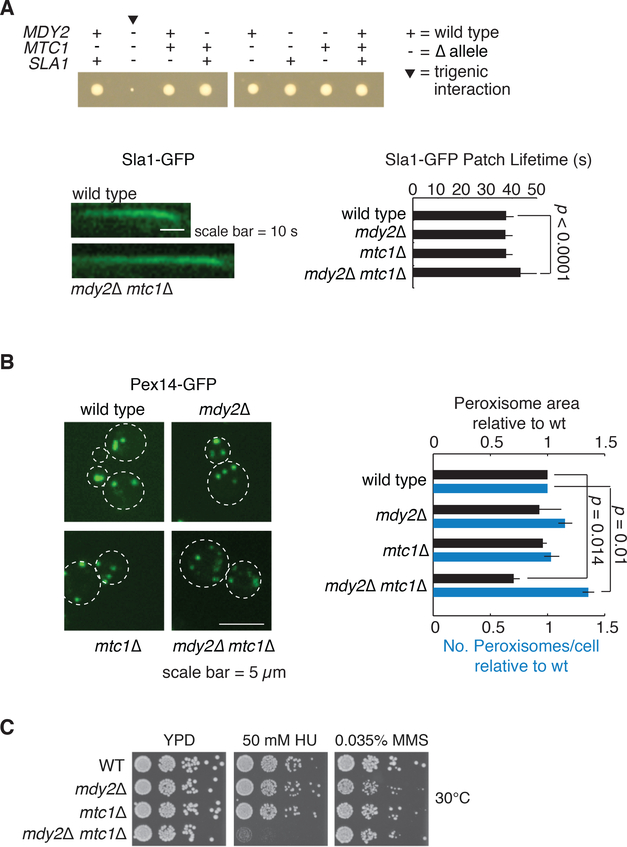
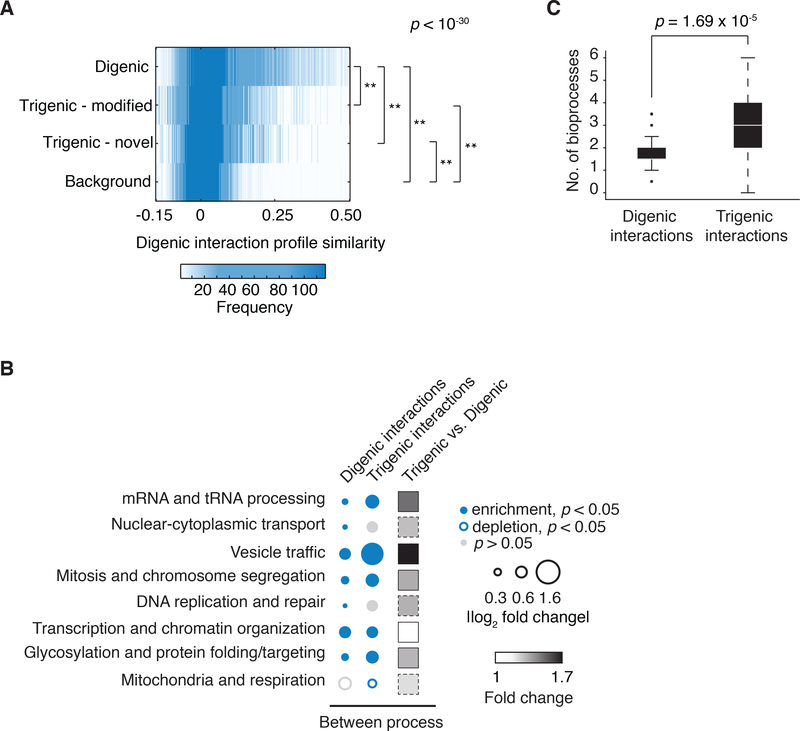
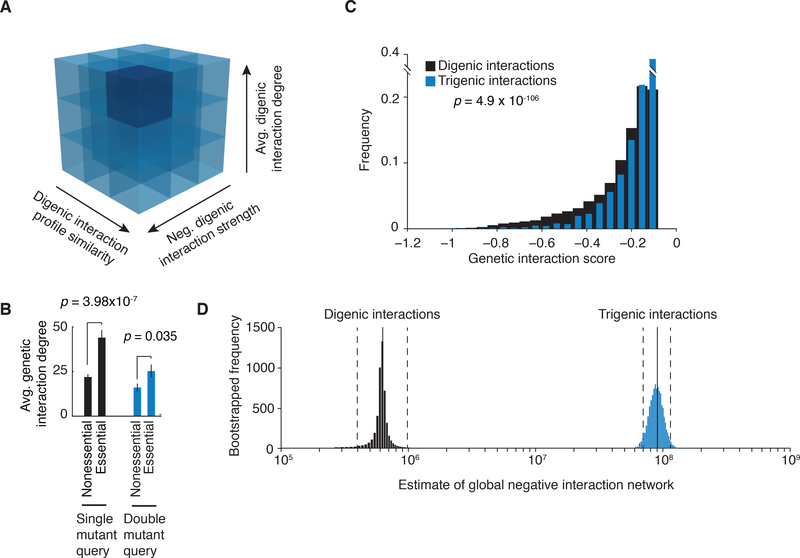
To systematically explore complex genetic interactions, we constructed ~200,000 yeast triple mutants and scored negative trigenic interactions. We selected double-mutant query genes across a broad spectrum of biological processes, spanning a range of quantitative features of the global digenic interaction network and tested for a genetic interaction with a third mutation. Trigenic interactions often occurred among functionally related genes, and essential genes were hubs on the trigenic network. Despite their functional enrichment, trigenic interactions tended to link genes in distant bioprocesses and displayed a weaker magnitude than digenic interactions. We estimate that the global trigenic interaction network is ~100 times as large as the global digenic network, highlighting the potential for complex genetic interactions to affect the biology of inheritance, including the genotype-to-phenotype relationship.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
If two deletions don't stop growth, try three.Science. 2018 Apr 20;360(6386):269-270. doi: 10.1126/science.aat4667. Science. 2018. PMID: 29674581 No abstract available.
-
Higher-order genetic interactions.Nat Methods. 2018 Jul;15(7):481. doi: 10.1038/s41592-018-0063-6. Nat Methods. 2018. PMID: 29967502 No abstract available.
References
-
- Hartman J. L. t., Garvik B, Hartwell L, Principles for the buffering of genetic variation. Science 291, 1001–1004 (2001). - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
