

Emergence of Vancomycin-Resistant Enterococcus faecium at an Australian Hospital: A Whole Genome Sequencing Analysis
- PMID: 29674657
- PMCID: PMC5908837
- DOI: 10.1038/s41598-018-24614-6
Emergence of Vancomycin-Resistant Enterococcus faecium at an Australian Hospital: A Whole Genome Sequencing Analysis
Abstract
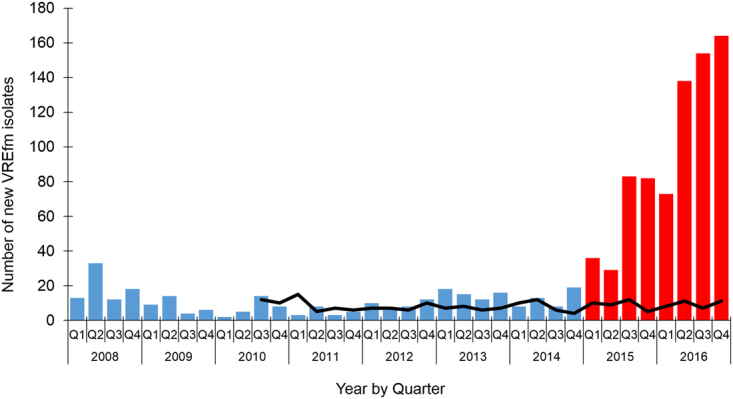
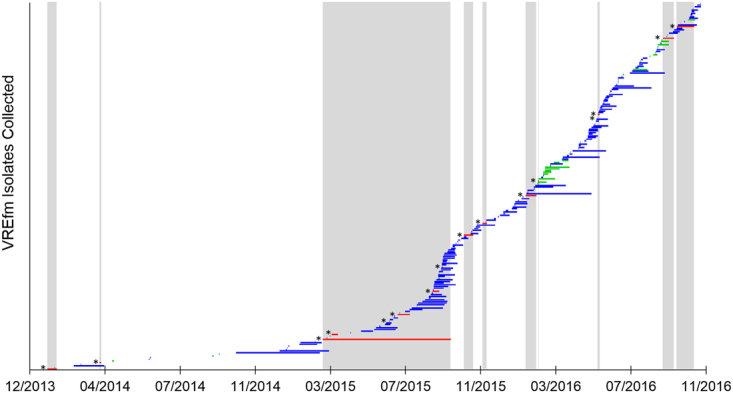
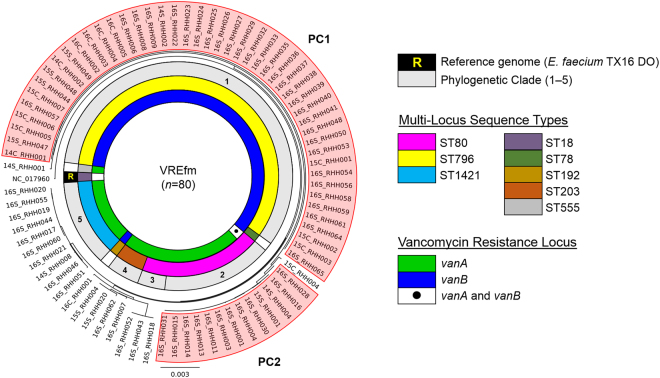
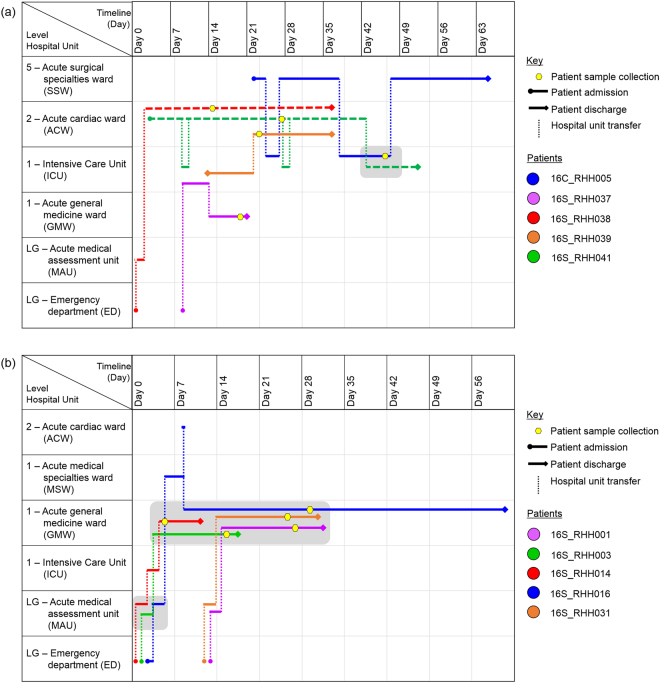
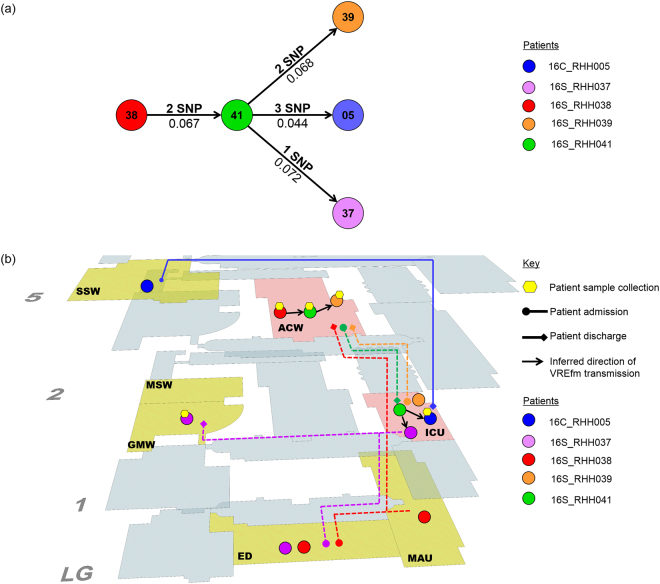
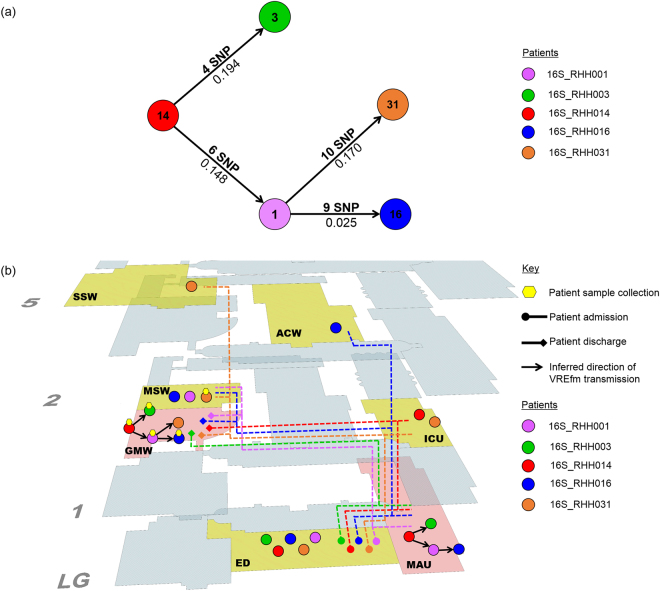
In 2015, a marked increase in vancomycin-resistant Enterococcus faecium (VREfm) isolation was detected at the Royal Hobart Hospital, Australia. The primary objective of this work was to examine the dynamics of VREfm transmission using whole genome data mapped to public health surveillance information. Screening and clinical isolates of VREfm from patients were typed for the specific vancomycin-resistance locus present. Of total isolates collected from 2014-2016 (n = 222), 15.3% and 84.7% harboured either the vanA or the vanB vancomycin-resistance locus, respectively. Whole-genome sequencing of 80 isolates was performed in conjunction with single-nucleotide polymorphic (SNP) analysis and in silico multi-locus sequence typing (MLST). Among the isolates sequenced, 5 phylogenetic clades were identified. The largest vanB clade belonged to MLST sequence type ST796 and contained clinical isolates from VREfm infections that clustered closely with isolates from colonised patients. Correlation of VREfm genotypes with spatio-temporal patient movements detected potential points of transmission within the hospital. ST80 emerged as the major vanA sequence type for which the most likely index case of a patient cluster was ascertained from SNP analyses. This work has identified the dominant clones associated with increased VREfm prevalence in a healthcare setting, and their likely direction of transmission.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- World Health Organisation. Report on the Burden of Endemic Health Care-Associated Infection Worldwide. (2011).
-
- Graves N, Halton K, Paterson D, Whitby M. Economic rationale for infection control in Australian hospitals. Healthcare Infection. 2009;14:81–88. doi: 10.1071/HI09010. - DOI
-
- Coombs, G. W. & Daley, D. A. Australian Enterococcal Sepsis Outcome Program (AESOP) 2016: Final Report. in AESOP - Australian Enterococcal Sepsis Outcome Program (2017).
-
- Australian Commission on Safety and Quality in Health Care (ACSQHC). AURA 2017: Second Australian report on antimicrobial use and resistance in human health. (Sydney, Australia, 2017).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
