

Hereditary renal cell carcinoma syndromes: diagnosis, surveillance and management
- PMID: 29680948
- PMCID: PMC6280834
- DOI: 10.1007/s00345-018-2288-5
Hereditary renal cell carcinoma syndromes: diagnosis, surveillance and management
Abstract
Purpose: Genetic factors have been implicated in the pathogenesis of renal cell carcinoma (RCC), with around 3% of cases having a family history. A greater knowledge of the genetics of inherited RCC has the potential to translate into novel therapeutic targets for sporadic RCC.
Methods: A literature review was performed summarising the current knowledge on hereditary RCC diagnosis, surveillance and management.
Results: Familial RCC is usually inherited in an autosomal dominant manner, although inherited RCC may present without a relevant family history. A number of familial RCC syndromes have been identified. Familial non-syndromic RCC is suspected when ≥ 2 relatives are affected in the absence of syndromic features, although clear diagnostic criteria are lacking. Young age at onset and bilateral/multicentric tumours are recognised characteristics which should prompt molecular genetic analysis. Surveillance in individuals at risk of inherited RCC aims to prevent morbidity and mortality via early detection of tumours. Though screening and management guidelines for some inherited RCC syndromes (e.g. von Hippel-Lindau disease, Birt-Hogg-Dube syndrome, hereditary leiomyomatosis) are well defined for rare cause of inherited RCC (e.g. germline BAP1 mutations), there is limited information regarding the lifetime RCC risks and the most appropriate screening modalities.
Conclusion: Increasing knowledge of the natural history and genetic basis has led to characterisation and tailored management of hereditary RCC syndromes. International data sharing of inherited RCC gene variant information may enable evidence-based improvements in the diagnosis, surveillance protocols and management of these rare conditions.
Keywords: Familial syndromic renal cancer; Genetics; von Hippel–Lindau review.
Conflict of interest statement
Conflict of interest
The author declares no conflict of interest.
Research involving human participants and/or animals
The following manuscript is a review of existing data. Therefore, this article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study (review) formal consent is not required.
Figures
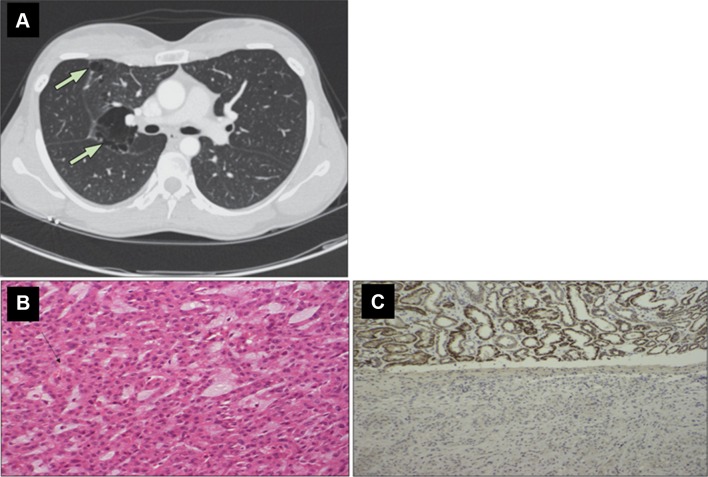
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
