

Stochastic processes constrain the within and between host evolution of influenza virus
- PMID: 29683424
- PMCID: PMC5933925
- DOI: 10.7554/eLife.35962
Stochastic processes constrain the within and between host evolution of influenza virus
Abstract
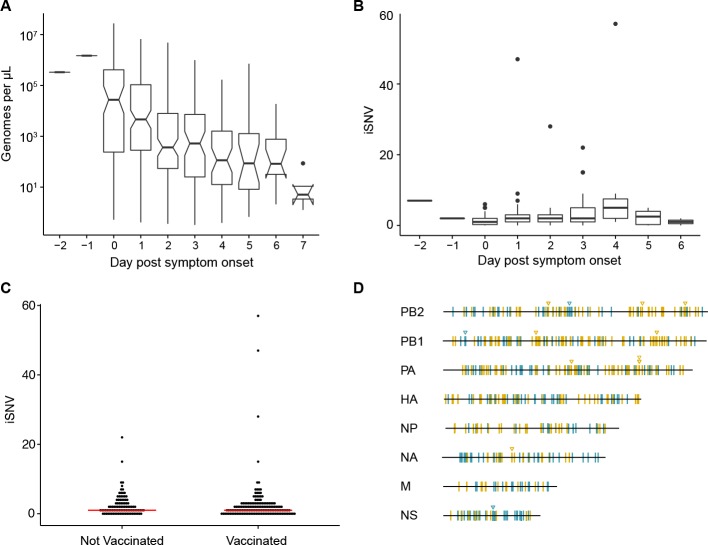
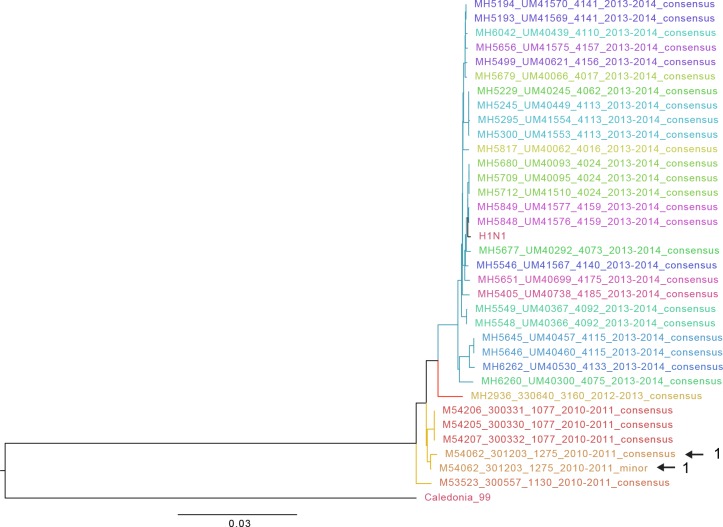
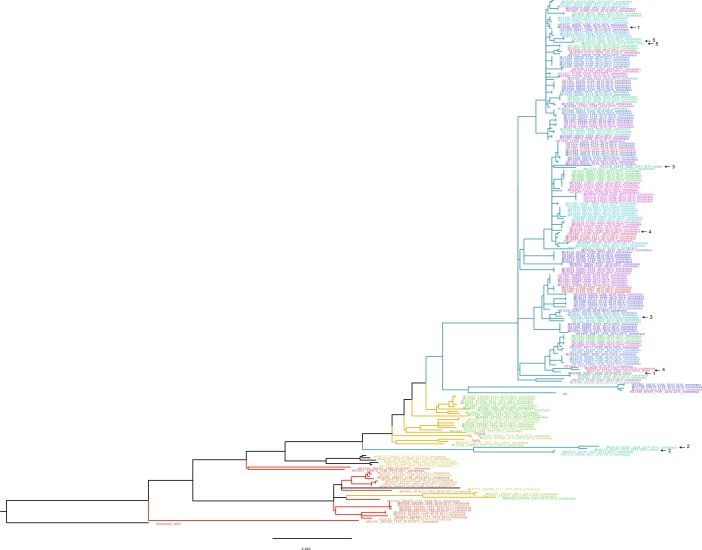
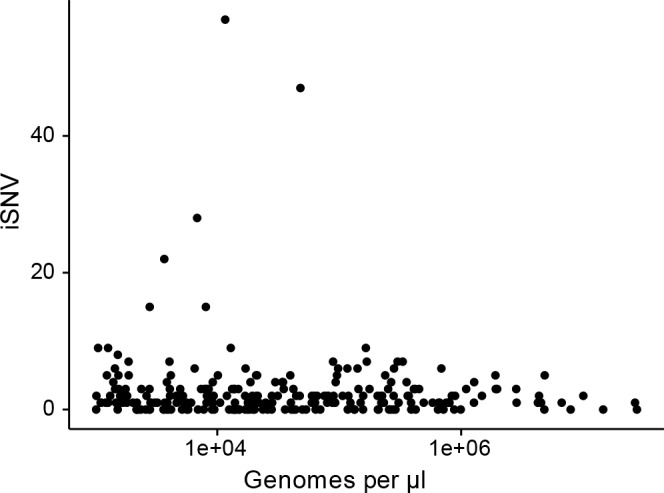
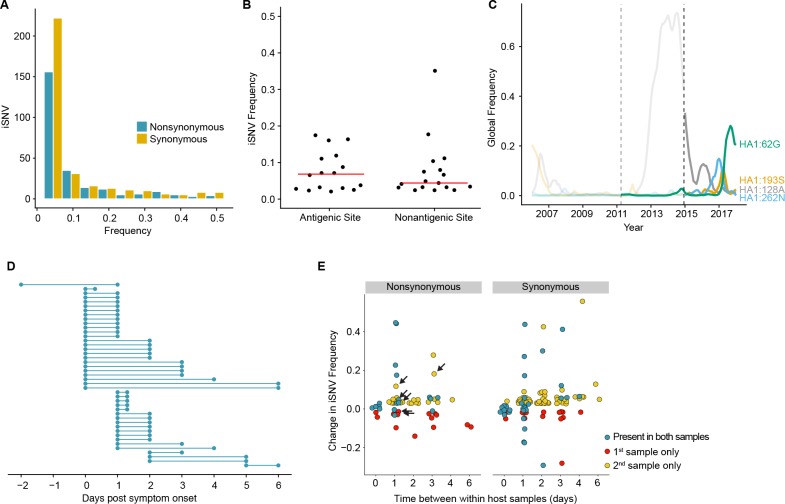
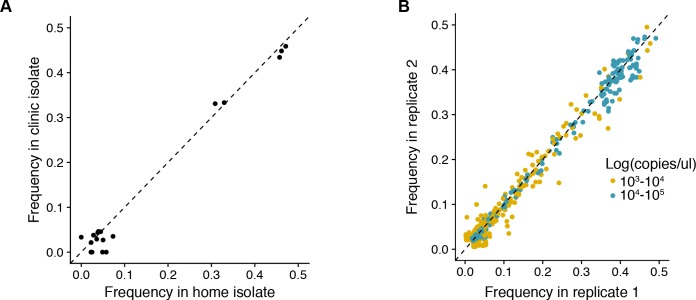
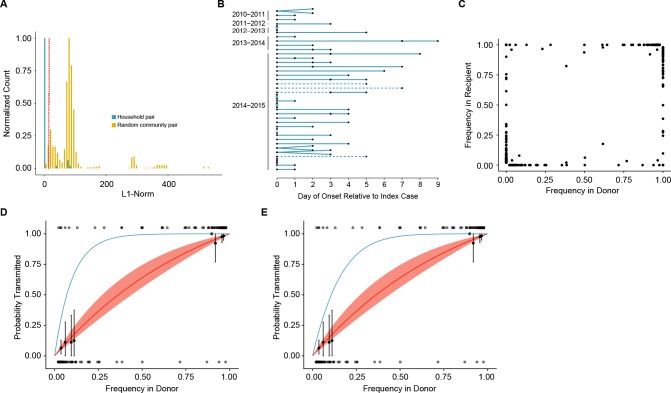
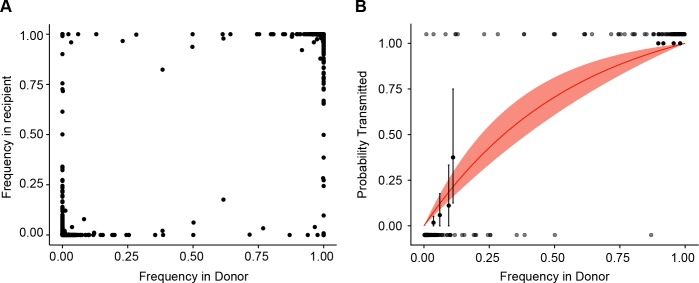
The evolutionary dynamics of influenza virus ultimately derive from processes that take place within and between infected individuals. Here we define influenza virus dynamics in human hosts through sequencing of 249 specimens from 200 individuals collected over 6290 person-seasons of observation. Because these viruses were collected from individuals in a prospective community-based cohort, they are broadly representative of natural infections with seasonal viruses. Consistent with a neutral model of evolution, sequence data from 49 serially sampled individuals illustrated the dynamic turnover of synonymous and nonsynonymous single nucleotide variants and provided little evidence for positive selection of antigenic variants. We also identified 43 genetically-validated transmission pairs in this cohort. Maximum likelihood optimization of multiple transmission models estimated an effective transmission bottleneck of 1-2 genomes. Our data suggest that positive selection is inefficient at the level of the individual host and that stochastic processes dominate the host-level evolution of influenza viruses.
Keywords: bottleneck; diversity; evolution; evolutionary biology; genomics; infectious disease; influenza virus; microbiology; transmission; virus.
© 2018, McCrone et al.
Conflict of interest statement
JM, RW, EM, RM, AM, AL No competing interests declared
Figures
References
-
- Bedford T, Riley S, Barr IG, Broor S, Chadha M, Cox NJ, Daniels RS, Gunasekaran CP, Hurt AC, Kelso A, Klimov A, Lewis NS, Li X, McCauley JW, Odagiri T, Potdar V, Rambaut A, Shu Y, Skepner E, Smith DJ, Suchard MA, Tashiro M, Wang D, Xu X, Lemey P, Russell CA. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature. 2015;523:217–220. doi: 10.1038/nature14460. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
