

Gut Microbiome Analysis Identifies Potential Etiological Factors in Acute Gastroenteritis
- PMID: 29685983
- PMCID: PMC6013661
- DOI: 10.1128/IAI.00060-18
Gut Microbiome Analysis Identifies Potential Etiological Factors in Acute Gastroenteritis
Abstract
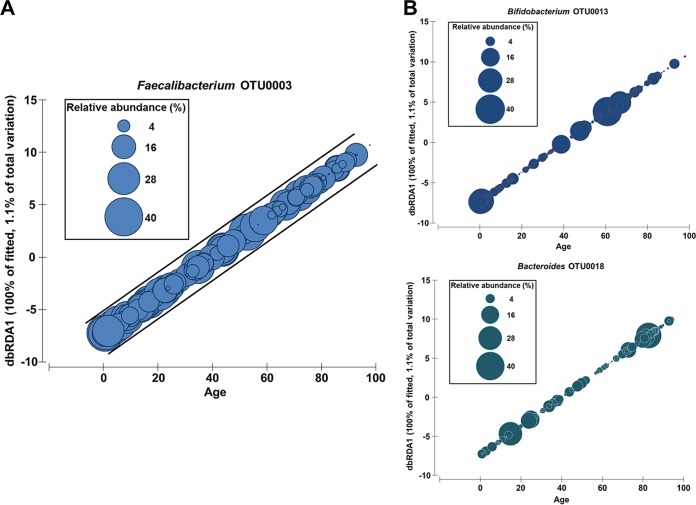
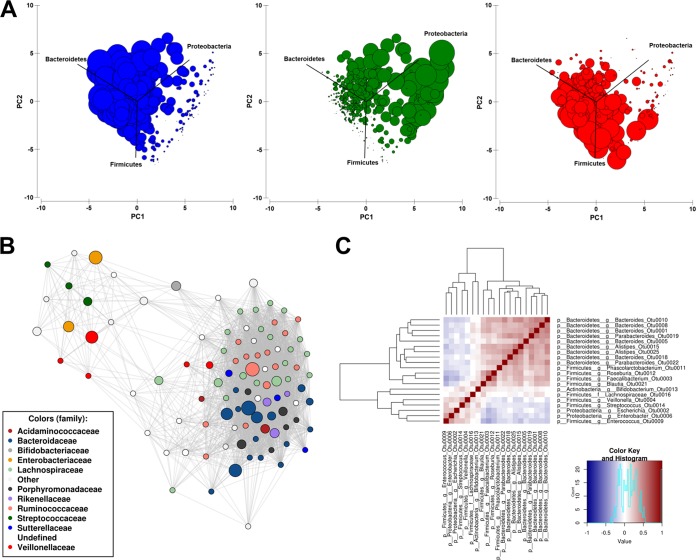
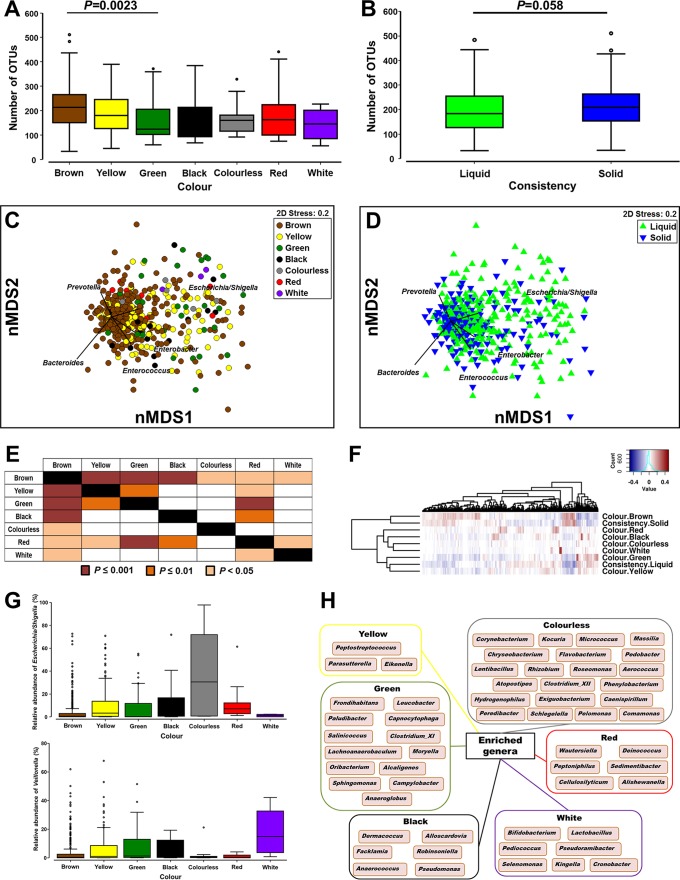
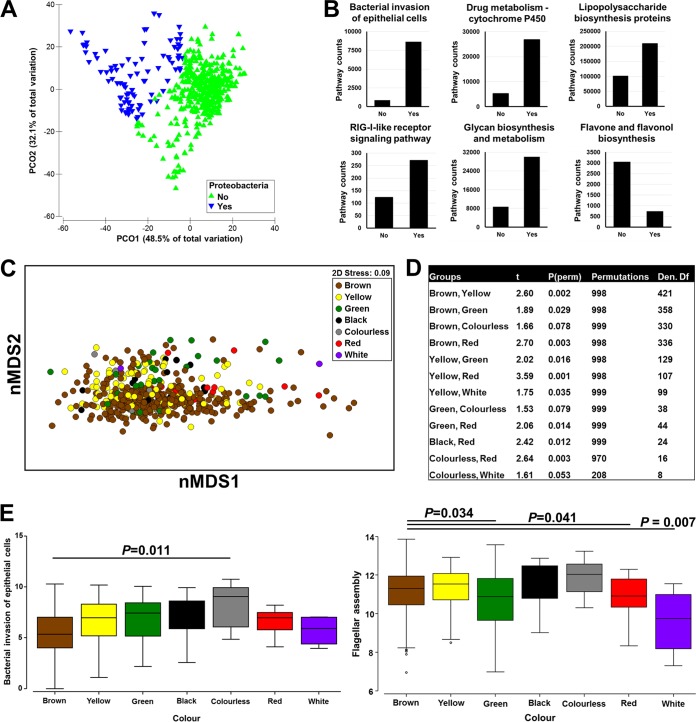
The morbidity and mortality resulting from acute gastroenteritis and associated chronic sequelae represent a substantial burden on health care systems worldwide. Few studies have investigated changes in the gut microbiome following an episode of acute gastroenteritis. By using nondirected 16S rRNA gene amplicon sequencing, the fecal microbiota of 475 patients with acute gastroenteritis was examined. Patient age was correlated with the overall microbial composition, with a decrease in the abundance of Faecalibacterium being observed in older patients. We observed the emergence of a potential Escherichia-Shigella-dominated enterotype in a subset of patients, and this enterotype was predicted to be more proinflammatory than the other common enterotypes, with the latter being dominated by Bacteroides or Faecalibacterium The increased abundance of Escherichia-Shigella did not appear to be associated with infection with an agent of a similar sequence similarity. Stool color and consistency were associated with the diversity and composition of the microbiome, with deviations from the norm (not brown and solid) showing increases in the abundances of bacteria such as Escherichia-Shigella and Veillonella Analysis of enriched outliers within the data identified a range of genera previously associated with gastrointestinal diseases, including Treponema, Proteus, Capnocytophaga, Arcobacter, Campylobacter, Haemophilus, Aeromonas, and Pseudomonas Our data represent the first in-depth analysis of gut microbiota in acute gastroenteritis. Phenotypic changes in stool color and consistency were associated with specific changes in the microbiota. Enriched bacterial taxa were detected in cases where no causative agent was identified by using routine diagnostic tests, suggesting that in the future, microbiome analyses may be utilized to improve diagnostics.
Keywords: Escherichia coli; acute gastroenteritis; enterotype; gut; microbiota; pathogenesis.
Copyright © 2018 American Society for Microbiology.
Figures
References
-
- World Health Organization. 2017. Diarrhoeal disease. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs330/en/ Accessed 28 October 2017.
-
- Kamdar K, Khakpour S, Chen J, Leone V, Brulc J, Mangatu T, Antonopoulos DA, Chang EB, Kahn SA, Kirschner BS, Young G, DePaolo RW. 2016. Genetic and metabolic signals during acute enteric bacterial infection alter the microbiota and drive progression to chronic inflammatory disease. Cell Host Microbe 19:21–31. doi: 10.1016/j.chom.2015.12.006. - DOI - PMC - PubMed
-
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Doré J, MetaHIT Consortium, Antolín M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, Denariaz G, et al. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180. doi: 10.1038/nature09944. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
