

Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma
- PMID: 29686388
- PMCID: PMC5934307
- DOI: 10.1038/s41588-018-0105-0
Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma
Abstract
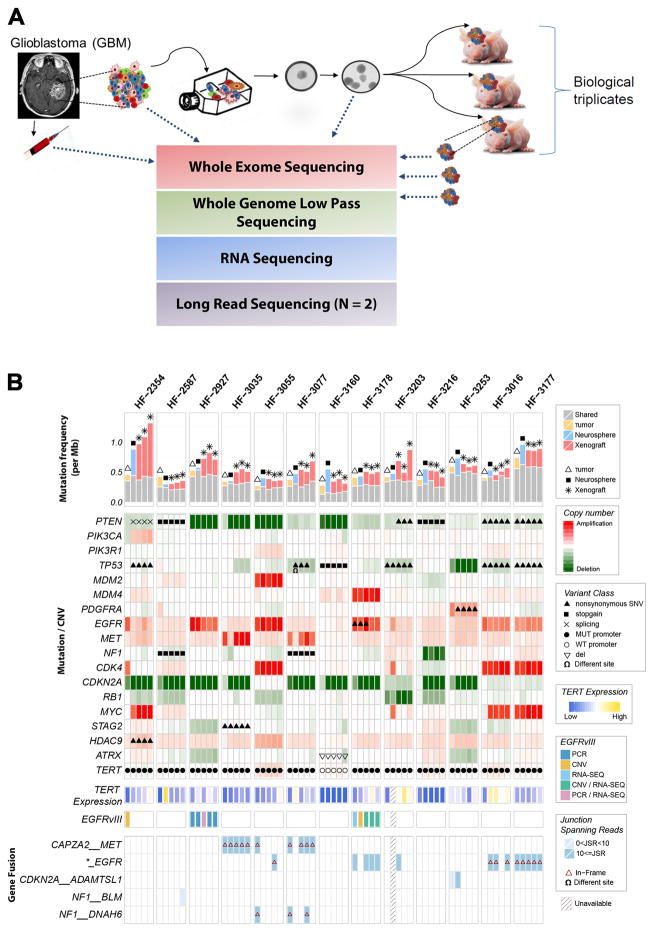
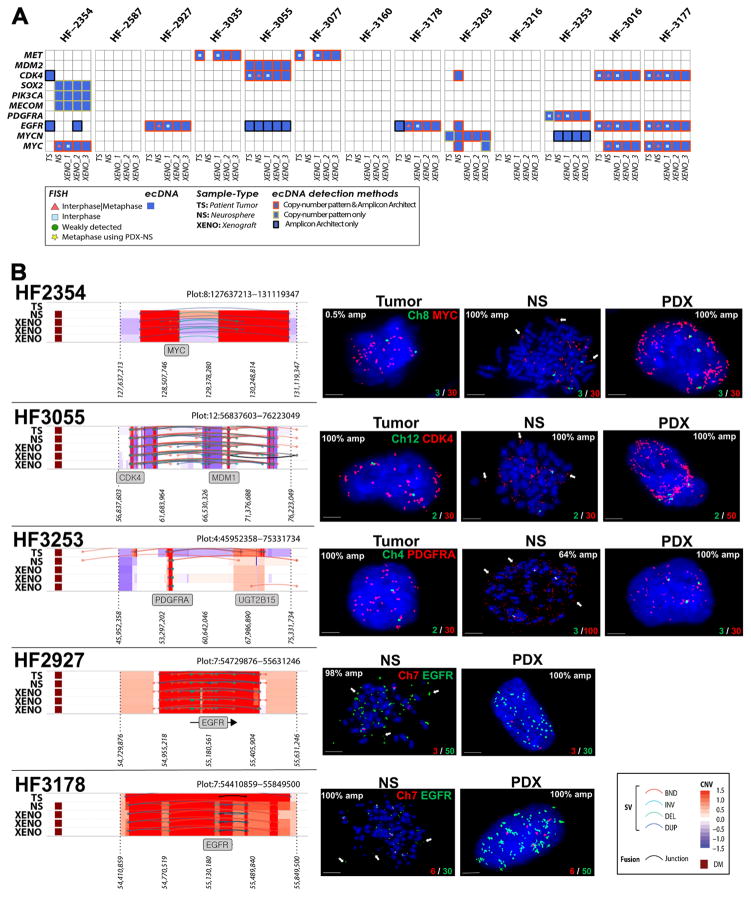
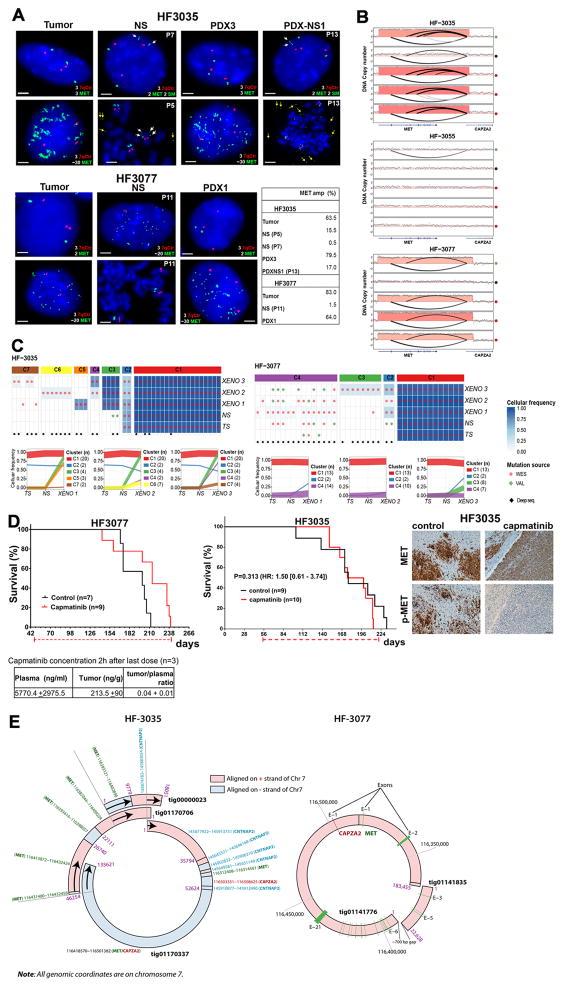
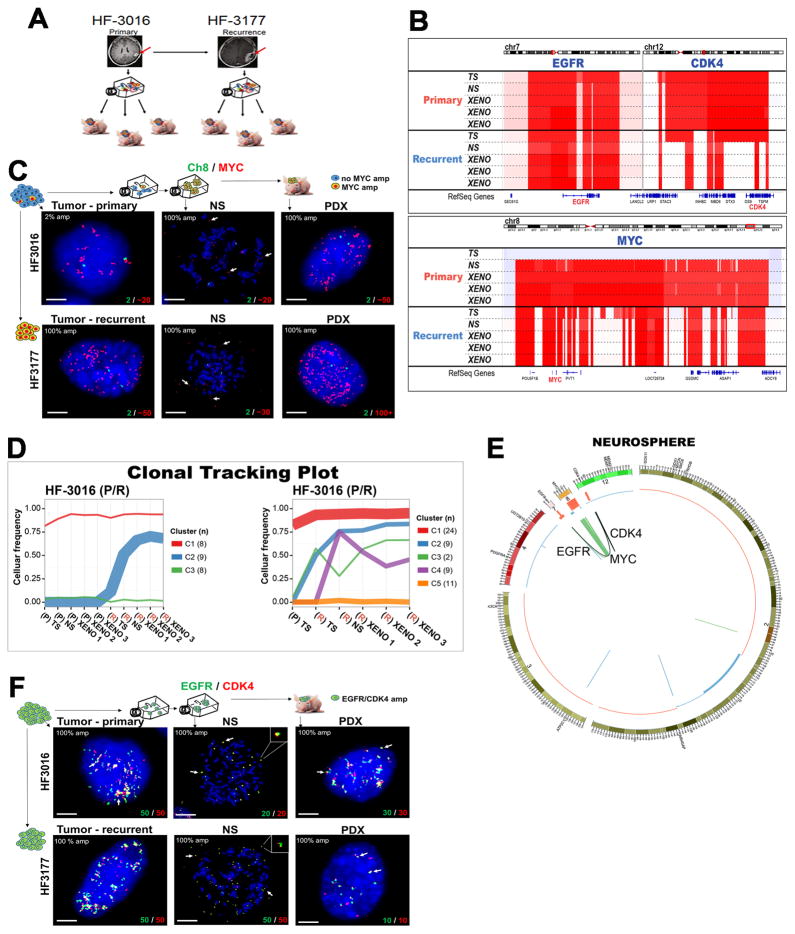
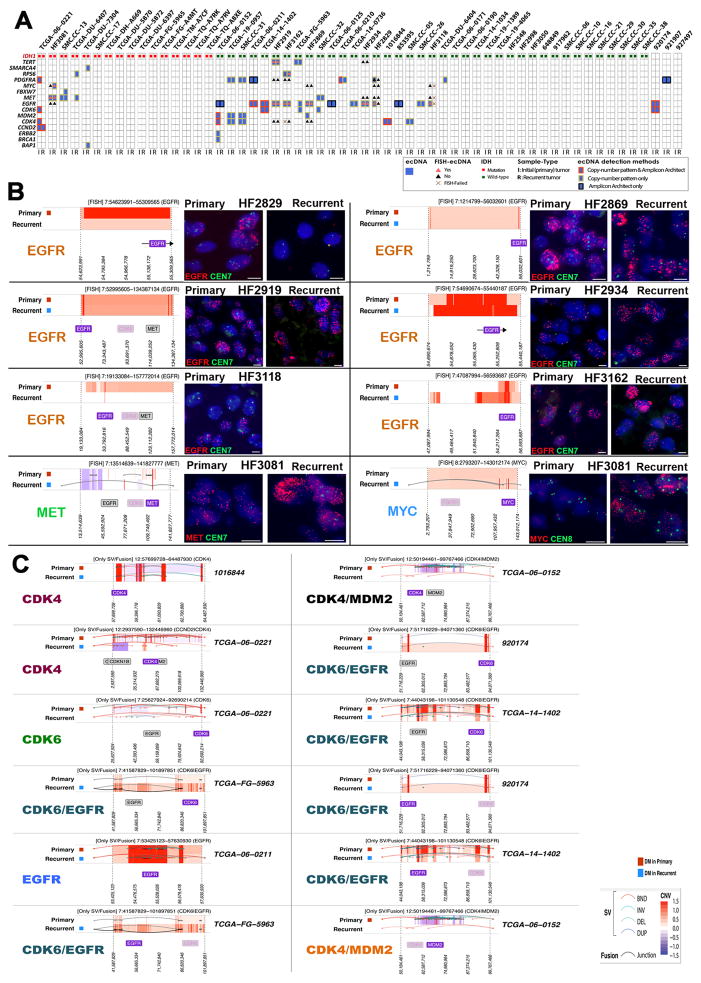
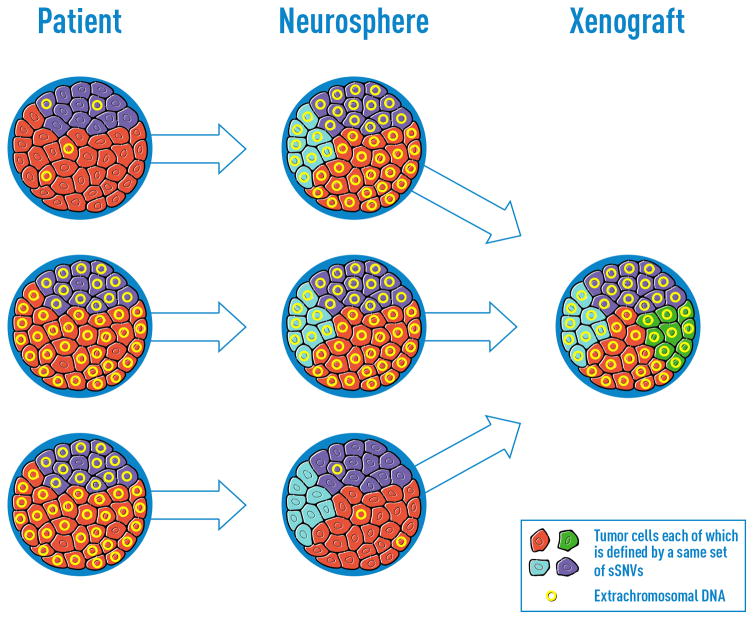
To understand how genomic heterogeneity of glioblastoma (GBM) contributes to poor therapy response, we performed DNA and RNA sequencing on GBM samples and the neurospheres and orthotopic xenograft models derived from them. We used the resulting dataset to show that somatic driver alterations including single-nucleotide variants, focal DNA alterations and oncogene amplification on extrachromosomal DNA (ecDNA) elements were in majority propagated from tumor to model systems. In several instances, ecDNAs and chromosomal alterations demonstrated divergent inheritance patterns and clonal selection dynamics during cell culture and xenografting. We infer that ecDNA was unevenly inherited by offspring cells, a characteristic that affects the oncogenic potential of cells with more or fewer ecDNAs. Longitudinal patient tumor profiling found that oncogenic ecDNAs are frequently retained throughout the course of disease. Our analysis shows that extrachromosomal elements allow rapid increase of genomic heterogeneity during GBM evolution, independently of chromosomal DNA alterations.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16:20–33. - PubMed
-
- Yap TA, Gerlinger M, Futreal PA, Pusztai L, Swanton C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012;4:127ps10. - PubMed
-
- Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
