

Transcript-indexed ATAC-seq for precision immune profiling
- PMID: 29686426
- PMCID: PMC5948148
- DOI: 10.1038/s41591-018-0008-8
Transcript-indexed ATAC-seq for precision immune profiling
Abstract
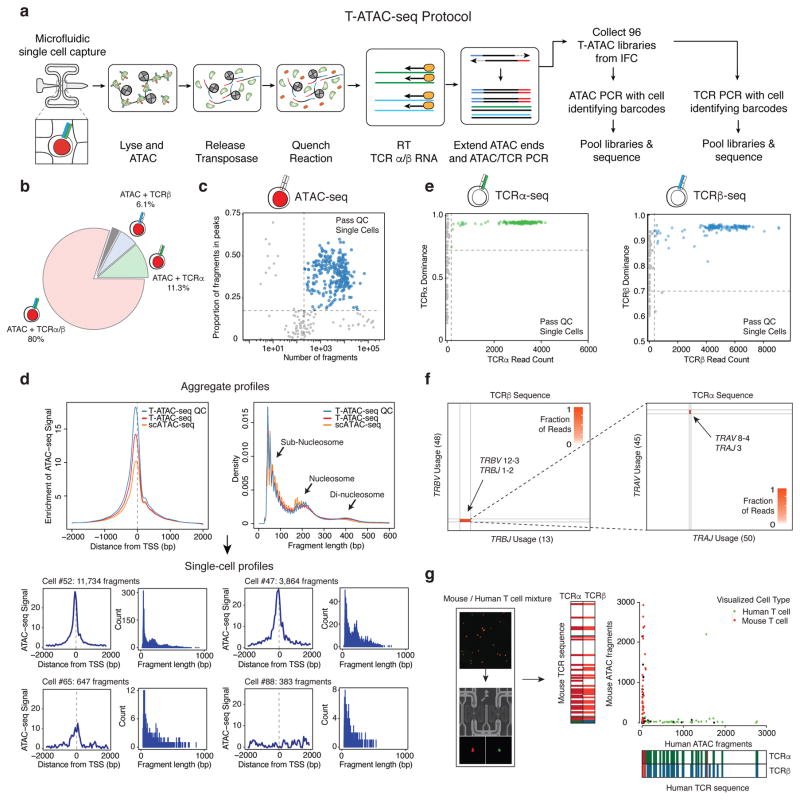
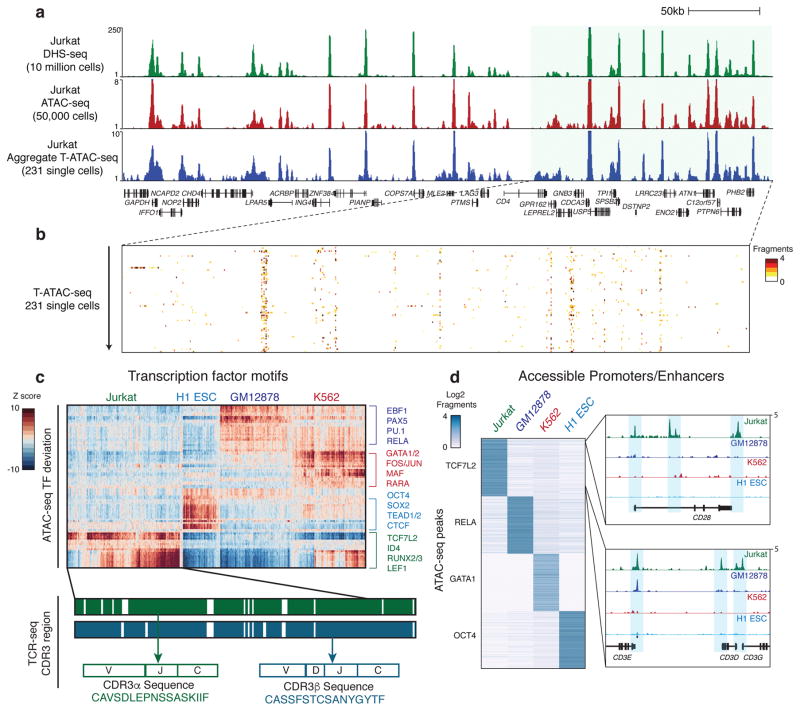
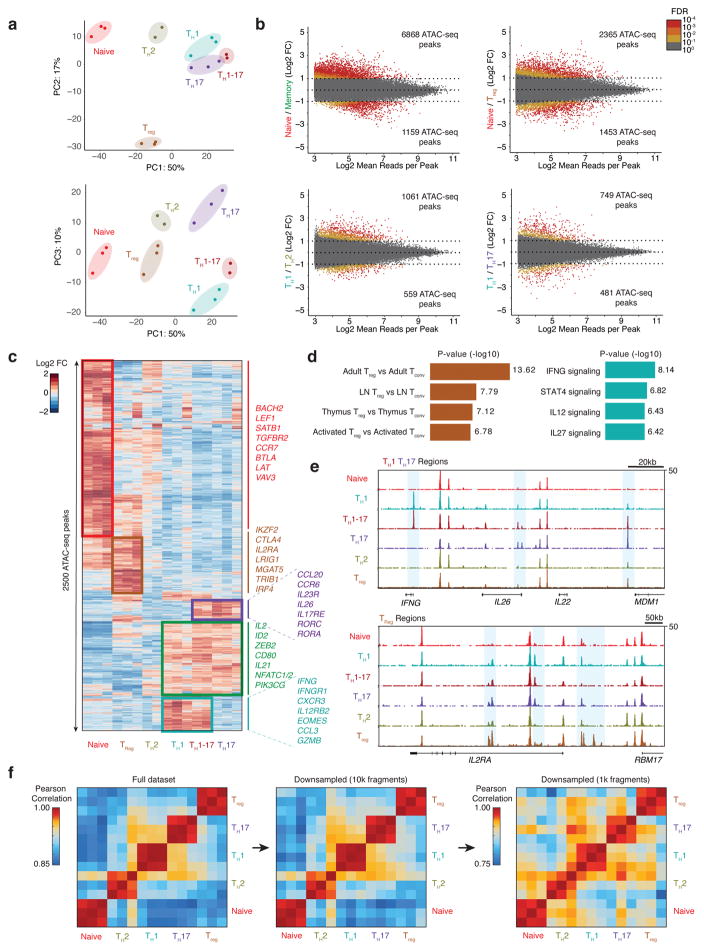
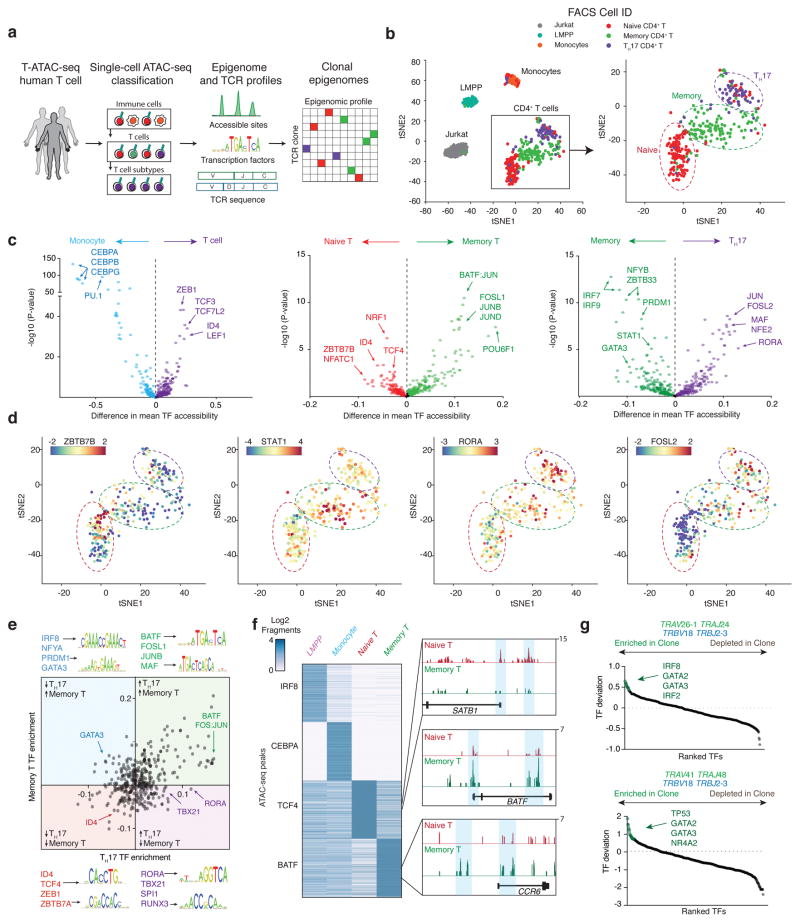
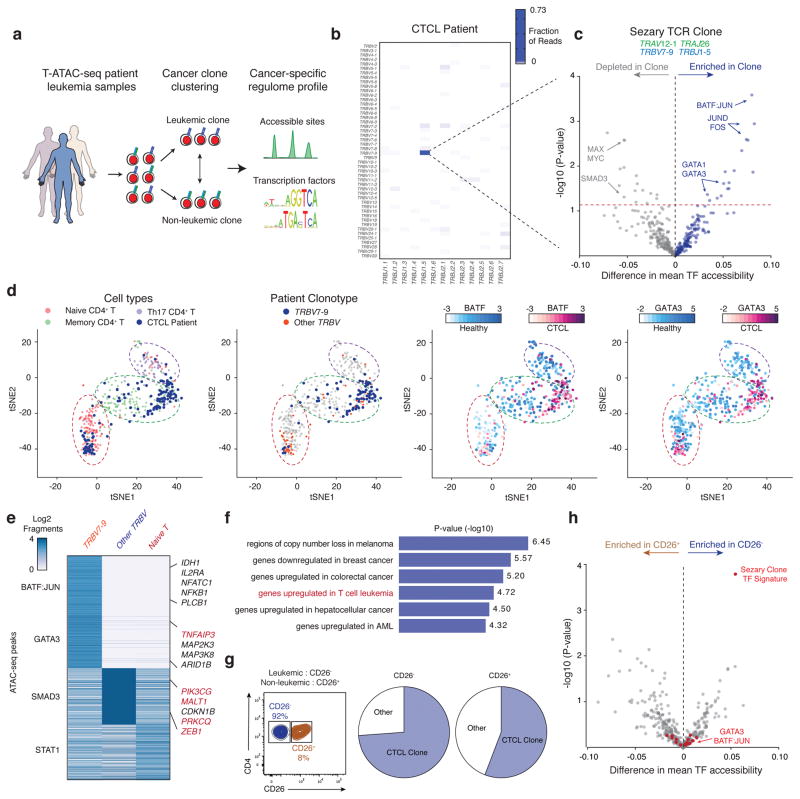
T cells create vast amounts of diversity in the genes that encode their T cell receptors (TCRs), which enables individual clones to recognize specific peptide-major histocompatibility complex (MHC) ligands. Here we combined sequencing of the TCR-encoding genes with assay for transposase-accessible chromatin with sequencing (ATAC-seq) analysis at the single-cell level to provide information on the TCR specificity and epigenomic state of individual T cells. By using this approach, termed transcript-indexed ATAC-seq (T-ATAC-seq), we identified epigenomic signatures in immortalized leukemic T cells, primary human T cells from healthy volunteers and primary leukemic T cells from patient samples. In peripheral blood CD4+ T cells from healthy individuals, we identified cis and trans regulators of naive and memory T cell states and found substantial heterogeneity in surface-marker-defined T cell populations. In patients with a leukemic form of cutaneous T cell lymphoma, T-ATAC-seq enabled identification of leukemic and nonleukemic regulatory pathways in T cells from the same individual by allowing separation of the signals that arose from the malignant clone from the background T cell noise. Thus, T-ATAC-seq is a new tool that enables analysis of epigenomic landscapes in clonal T cells and should be valuable for studies of T cell malignancy, immunity and immunotherapy.
Conflict of interest statement
H.Y.C. and W.J.G. are founders of Epinomics and members of its scientific advisory board. H.Y.C. is a founder of Accent Therapeutics and a member of its scientific advisory board. H.Y.C. is a member of the scientific advisory board of Spring Discovery.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
