

Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2
- PMID: 29690653
- PMCID: PMC5979509
- DOI: 10.3390/ijms19041264
Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2
Abstract
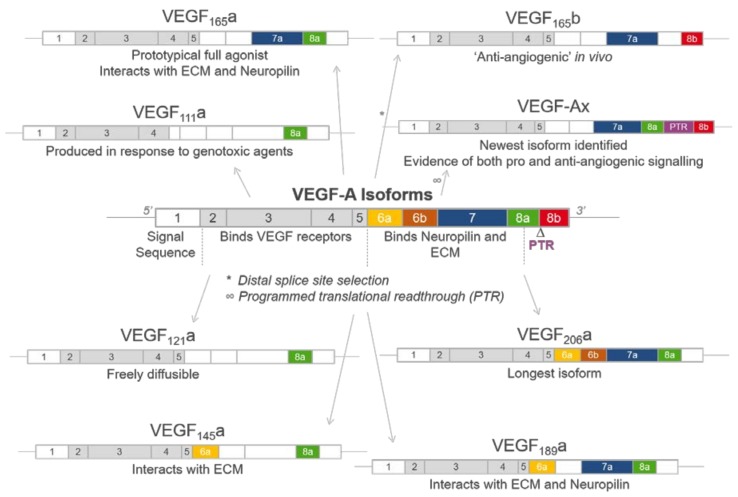
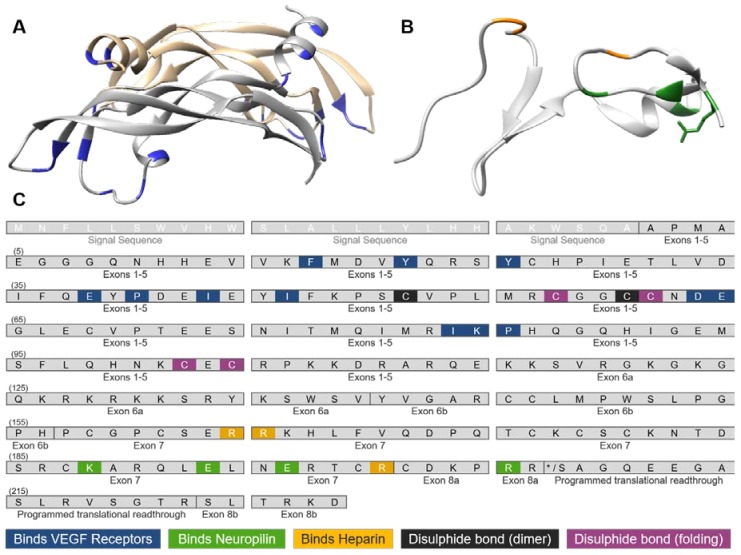
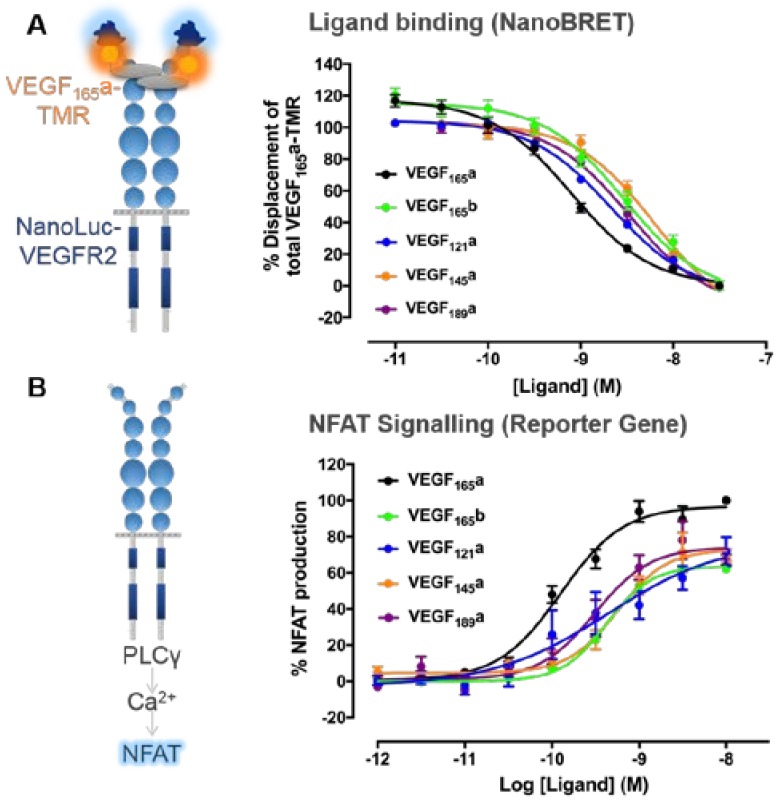
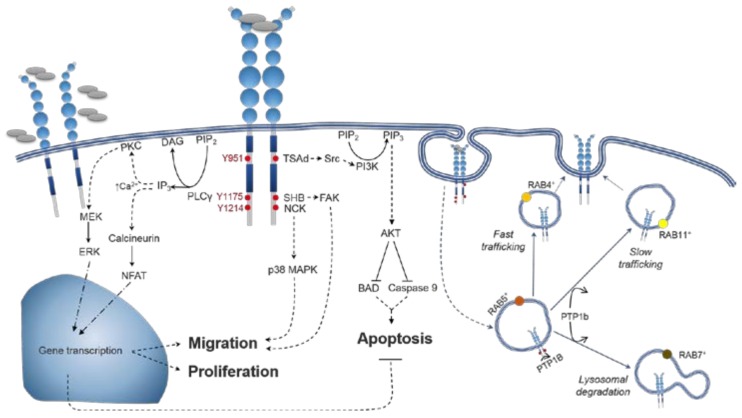
Vascular endothelial growth factor-A (VEGF-A) is a key mediator of angiogenesis, signalling via the class IV tyrosine kinase receptor family of VEGF Receptors (VEGFRs). Although VEGF-A ligands bind to both VEGFR1 and VEGFR2, they primarily signal via VEGFR2 leading to endothelial cell proliferation, survival, migration and vascular permeability. Distinct VEGF-A isoforms result from alternative splicing of the Vegfa gene at exon 8, resulting in VEGFxxxa or VEGFxxxb isoforms. Alternative splicing events at exons 5⁻7, in addition to recently identified posttranslational read-through events, produce VEGF-A isoforms that differ in their bioavailability and interaction with the co-receptor Neuropilin-1. This review explores the molecular pharmacology of VEGF-A isoforms at VEGFR2 in respect to ligand binding and downstream signalling. To understand how VEGF-A isoforms have distinct signalling despite similar affinities for VEGFR2, this review re-evaluates the typical classification of these isoforms relative to the prototypical, “pro-angiogenic” VEGF165a. We also examine the molecular mechanisms underpinning the regulation of VEGF-A isoform signalling and the importance of interactions with other membrane and extracellular matrix proteins. As approved therapeutics targeting the VEGF-A/VEGFR signalling axis largely lack long-term efficacy, understanding these isoform-specific mechanisms could aid future drug discovery efforts targeting VEGF receptor pharmacology.
Keywords: angiogenesis; blood vessel; endothelial cells; receptor tyrosine kinase inhibitors; splicing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
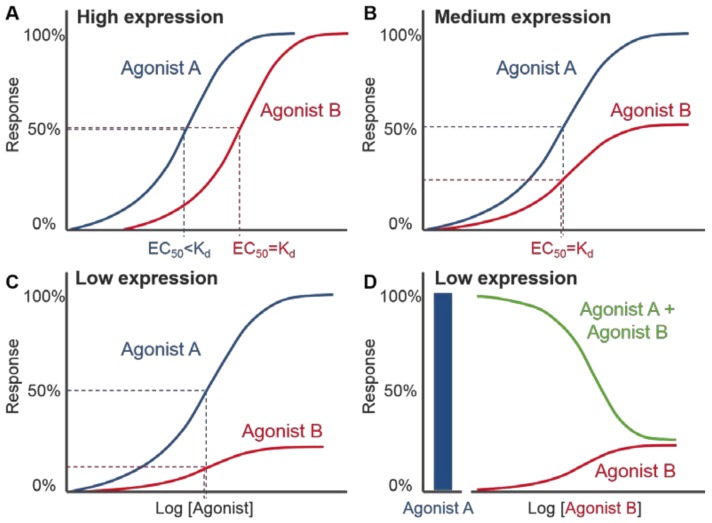
References
-
- Tepper O.M., Capla J.M., Galiano R.D., Ceradini D.J., Callaghan M.J., Kleinman M.E., Gurtner G.C. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood. 2005;105:1068–1077. doi: 10.1182/blood-2004-03-1051. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
