

Cross-species inference of long non-coding RNAs greatly expands the ruminant transcriptome
- PMID: 29690875
- PMCID: PMC5926538
- DOI: 10.1186/s12711-018-0391-0
Cross-species inference of long non-coding RNAs greatly expands the ruminant transcriptome
Abstract
Background: mRNA-like long non-coding RNAs (lncRNAs) are a significant component of mammalian transcriptomes, although most are expressed only at low levels, with high tissue-specificity and/or at specific developmental stages. Thus, in many cases lncRNA detection by RNA-sequencing (RNA-seq) is compromised by stochastic sampling. To account for this and create a catalogue of ruminant lncRNAs, we compared de novo assembled lncRNAs derived from large RNA-seq datasets in transcriptional atlas projects for sheep and goats with previous lncRNAs assembled in cattle and human. We then combined the novel lncRNAs with the sheep transcriptional atlas to identify co-regulated sets of protein-coding and non-coding loci.
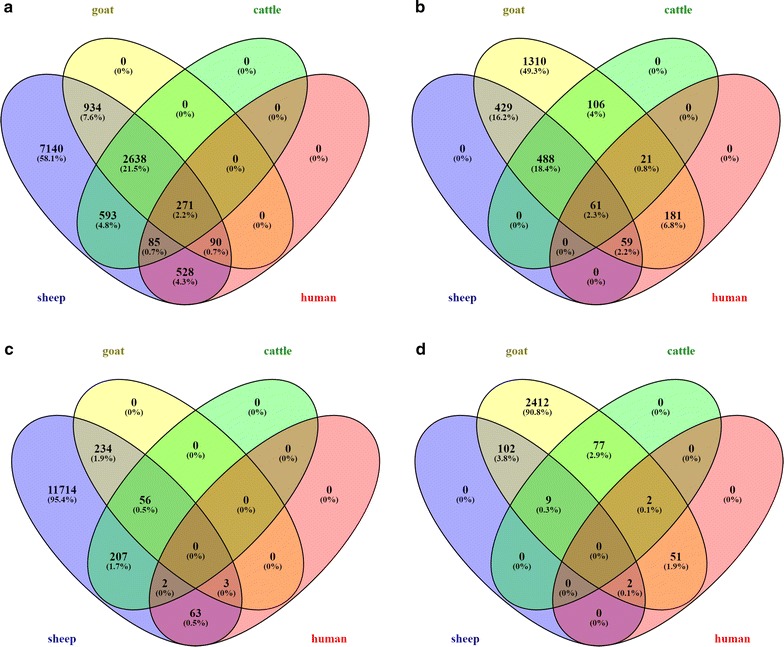
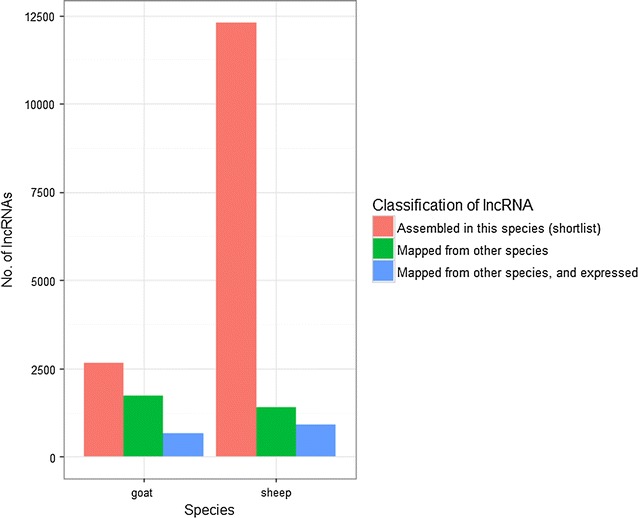
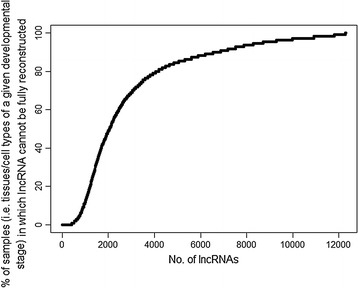
Results: Few lncRNAs could be reproducibly assembled from a single dataset, even with deep sequencing of the same tissues from multiple animals. Furthermore, there was little sequence overlap between lncRNAs that were assembled from pooled RNA-seq data. We combined positional conservation (synteny) with cross-species mapping of candidate lncRNAs to identify a consensus set of ruminant lncRNAs and then used the RNA-seq data to demonstrate detectable and reproducible expression in each species. In sheep, 20 to 30% of lncRNAs were located close to protein-coding genes with which they are strongly co-expressed, which is consistent with the evolutionary origin of some ncRNAs in enhancer sequences. Nevertheless, most of the lncRNAs are not co-expressed with neighbouring protein-coding genes.
Conclusions: Alongside substantially expanding the ruminant lncRNA repertoire, the outcomes of our analysis demonstrate that stochastic sampling can be partly overcome by combining RNA-seq datasets from related species. This has practical implications for the future discovery of lncRNAs in other species.
Figures

References
-
- Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. - PubMed
-
- Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016;17:756–770. - PubMed
-
- Chen J, Xue Y. Emerging roles of non-coding RNAs in epigenetic regulation. Sci China Life Sci. 2016;59:227–235. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- BB/L001209/1/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/P013732/1/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/D/20211552/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/J004243/1/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/D/2021550/Biotechnology and Biological Sciences Research Council (GB)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
