

Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese
- PMID: 29691385
- PMCID: PMC5915442
- DOI: 10.1038/s41467-018-03274-0
Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese
Abstract
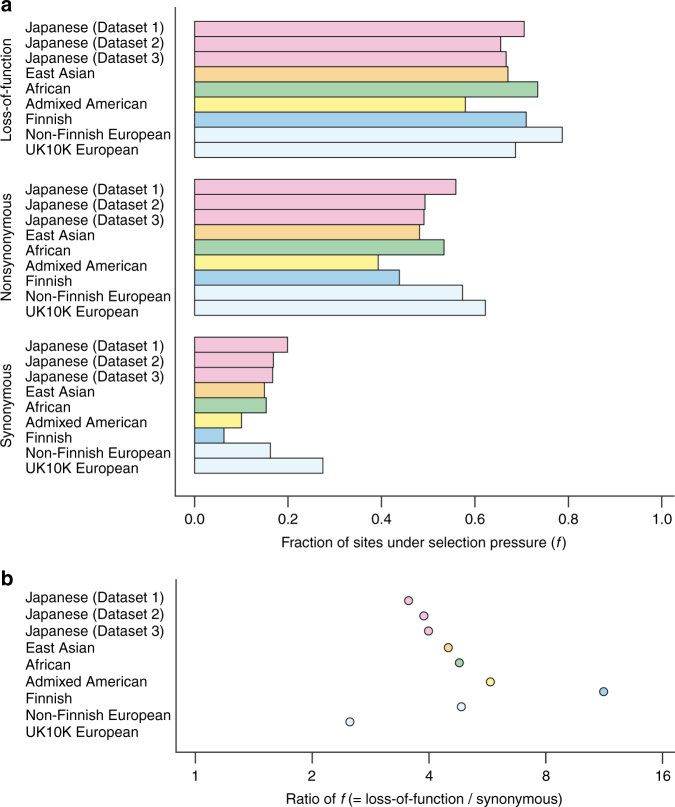
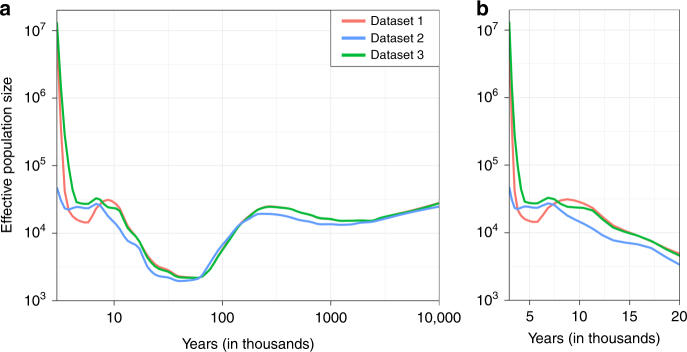
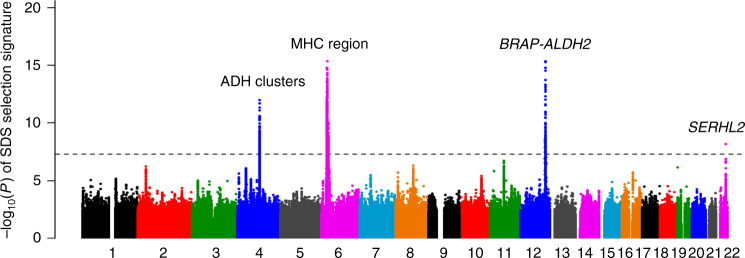
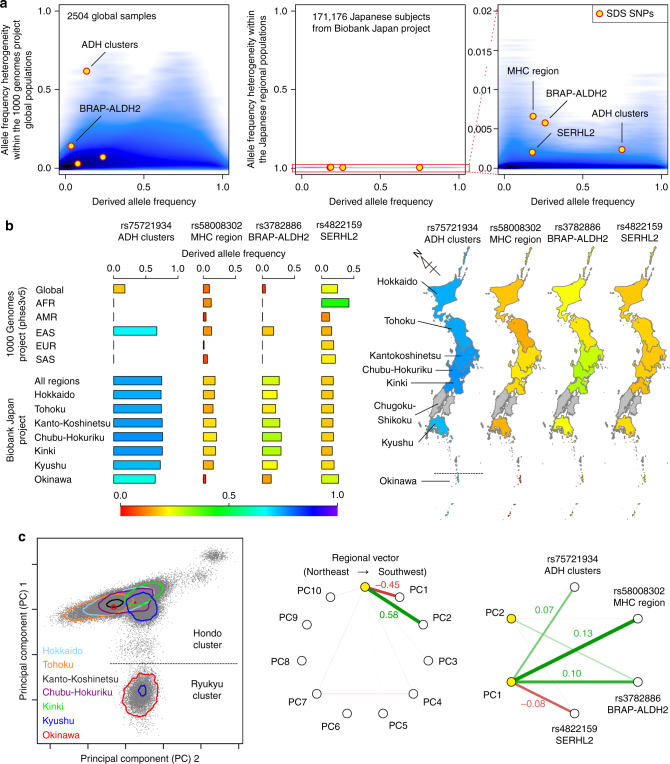
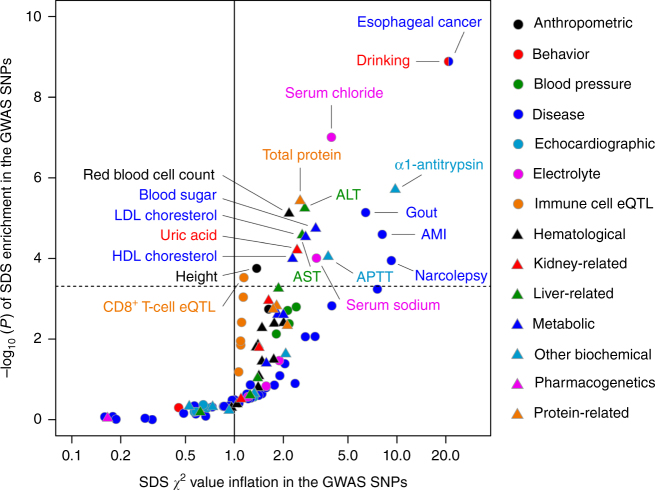
Understanding natural selection is crucial to unveiling evolution of modern humans. Here, we report natural selection signatures in the Japanese population using 2234 high-depth whole-genome sequence (WGS) data (25.9×). Using rare singletons, we identify signals of very recent selection for the past 2000-3000 years in multiple loci (ADH cluster, MHC region, BRAP-ALDH2, SERHL2). In large-scale genome-wide association study (GWAS) dataset (n = 171,176), variants with selection signatures show enrichment in heterogeneity of derived allele frequency spectra among the geographic regions of Japan, highlighted by two major regional clusters (Hondo and Ryukyu). While the selection signatures do not show enrichment in archaic hominin-derived genome sequences, they overlap with the SNPs associated with the modern human traits. The strongest overlaps are observed for the alcohol or nutrition metabolism-related traits. Our study illustrates the value of high-depth WGS to understand evolution and their relationship with disease risk.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
