

De novo main-chain modeling for EM maps using MAINMAST
- PMID: 29691408
- PMCID: PMC5915429
- DOI: 10.1038/s41467-018-04053-7
De novo main-chain modeling for EM maps using MAINMAST
Abstract
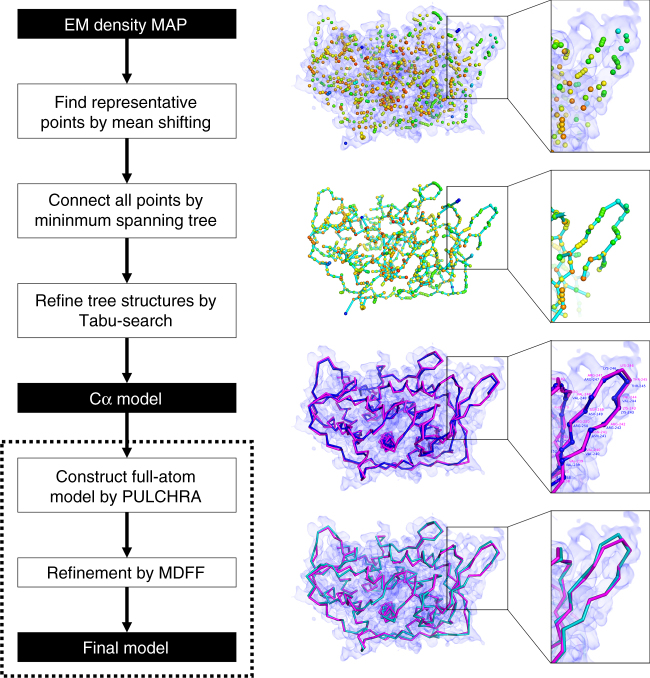
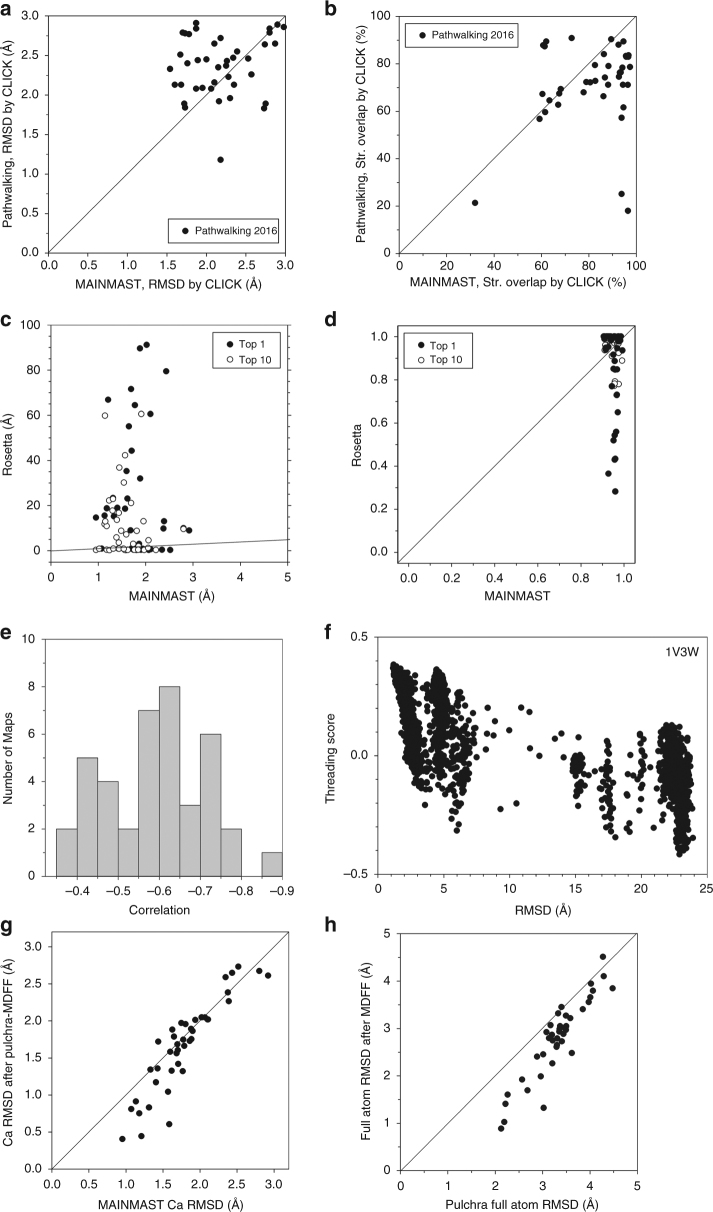
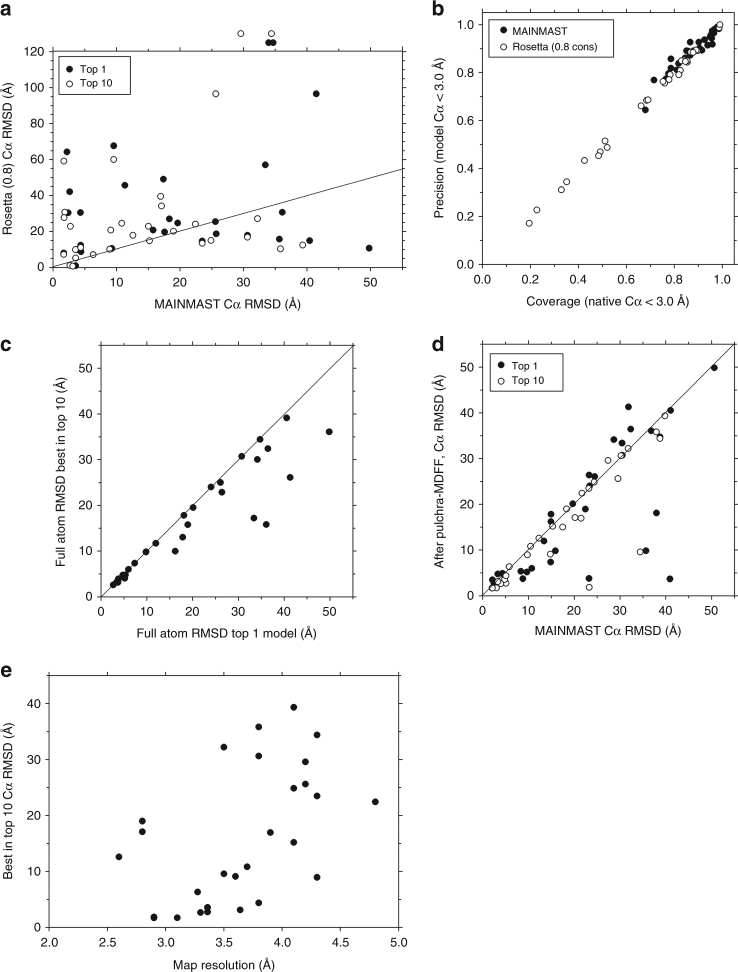
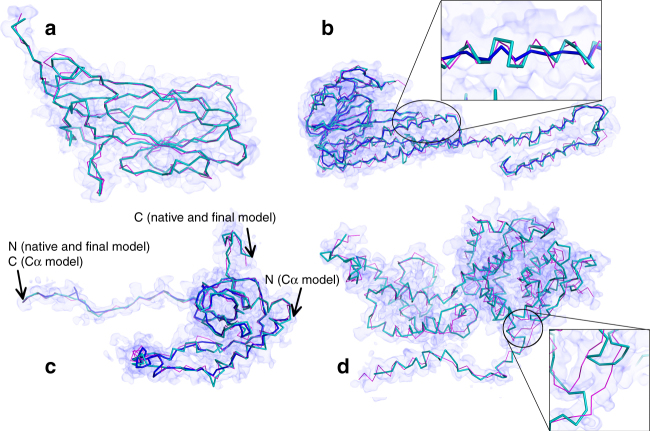
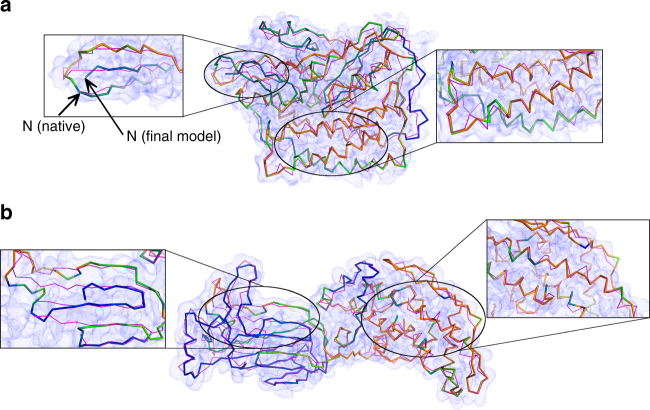
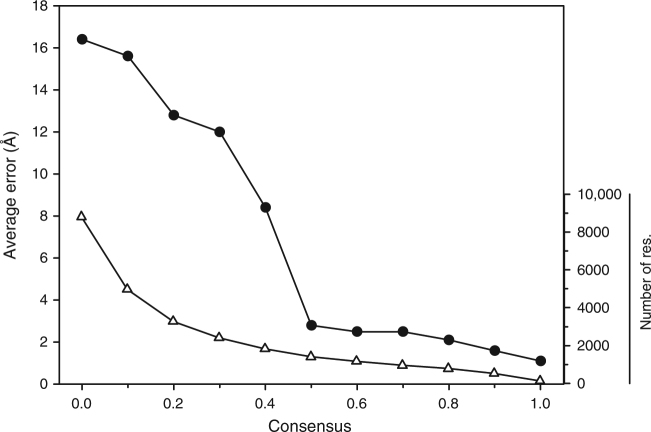
An increasing number of protein structures are determined by cryo-electron microscopy (cryo-EM) at near atomic resolution. However, tracing the main-chains and building full-atom models from EM maps of ~4-5 Å is still not trivial and remains a time-consuming task. Here, we introduce a fully automated de novo structure modeling method, MAINMAST, which builds three-dimensional models of a protein from a near-atomic resolution EM map. The method directly traces the protein's main-chain and identifies Cα positions as tree-graph structures in the EM map. MAINMAST performs significantly better than existing software in building global protein structure models on data sets of 40 simulated density maps at 5 Å resolution and 30 experimentally determined maps at 2.6-4.8 Å resolution. In another benchmark of building missing fragments in protein models for EM maps, MAINMAST builds fragments of 11-161 residues long with an average RMSD of 2.68 Å.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
