

An estimation of the prevalence of genomic disorders using chromosomal microarray data
- PMID: 29691480
- PMCID: PMC6019170
- DOI: 10.1038/s10038-018-0451-x
An estimation of the prevalence of genomic disorders using chromosomal microarray data
Abstract
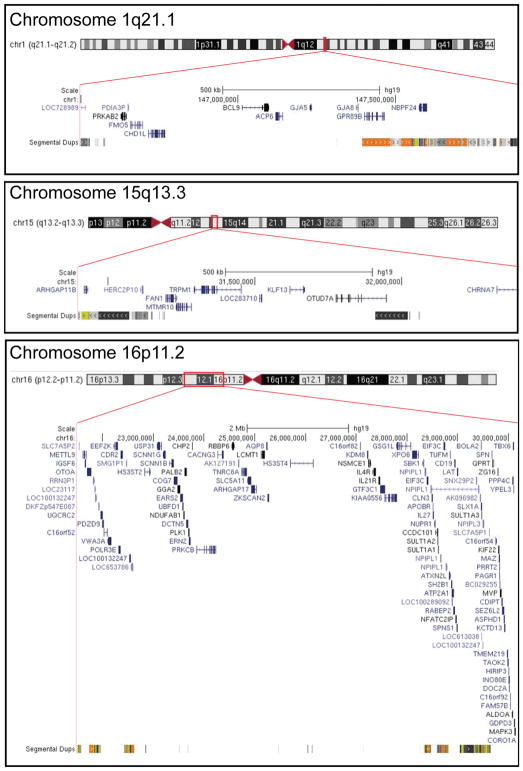
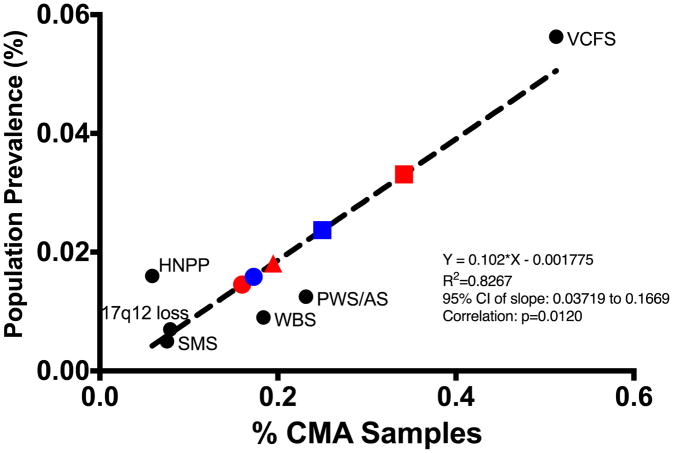
Multiple genomic disorders result from recurrent deletions or duplications between low copy repeat (LCR) clusters, mediated by nonallelic homologous recombination. These copy number variants (CNVs) often exhibit variable expressivity and/or incomplete penetrance. However, the population prevalence of many genomic disorders has not been estimated accurately. A subset of genomic disorders similarly characterized by CNVs between LCRs have been studied epidemiologically, including Williams-Beuren syndrome (7q11.23), Smith-Magenis syndrome (17p11.2), velocardiofacial syndrome (22q11.21), Prader-Willi/Angelman syndromes (15q11.2q12), 17q12 deletion syndrome, and Charcot-Marie-Tooth neuropathy type 1/hereditary neuropathy with liability to pressure palsy (PMP22, 17q11.2). We have generated a method to estimate prevalence of highly penetrant genomic disorders by (1) leveraging epidemiological data for genomic disorders with previously reported prevalence estimates, (2) obtaining chromosomal microarray data on genomic disorders from a large medical genetics clinic; and (3) utilizing these in a linear regression model to determine the prevalence of this syndromic copy number change among the general population. Using our algorithm, the prevalence for five clinically relevant recurrent genomic disorders: 1q21.1 microdeletion (1/6882 live births) and microduplication syndromes (1/6309), 15q13.3 microdeletion syndrome (1/5525), and 16p11.2 microdeletion (1/3021) and microduplication syndromes (1/4216), were determined. These findings will inform epidemiological strategies for evaluating those conditions, and our method may be useful to evaluate the prevalence of other highly penetrant genomic disorders.
Conflict of interest statement
None of the authors have a conflict of interest to declare.
Figures
References
-
- Miller D, Adam M, Aradhya S, Biesecker L, Brothman A, Carter N, Church D, Crolla J, Eichler E, Epstein C, et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. The American Journal of Human Genetics. 2010;86:749–764. - PMC - PubMed
-
- Õiglane-Shlik E, Talvik T, Žordania R, Põder H, Kahre T, Raukas E, Ilus T, Tasa G, Bartsch O, Väisänen M, et al. Prevalence of Angelman syndrome and Prader–Willi syndrome in Estonian children: Sister syndromes not equally represented. Am J Med Genet A. 2006;140A:1936–1943. - PubMed
-
- Kyllerman On the prevalence of Angelman syndrome. Am J Med Genet. 1995;59:405–405. - PubMed
-
- Meretoja P, Silander K, Kalimo H, Aula P, Meretoja A, Savontaus ML. Epidemiology of hereditary neuropathy with liability to pressure palsies (HNPP) in south western Finland. Neuromuscul Disord. 1997;7:529–532. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical