

Review
doi: 10.1155/2018/4258387.
eCollection 2018.
Modelling Cooperative Tumorigenesis in Drosophila
Affiliations
- PMID: 29693007
- PMCID: PMC5859872
- DOI: 10.1155/2018/4258387
Item in Clipboard
Review
Modelling Cooperative Tumorigenesis in Drosophila
Biomed Res Int.
.
Abstract
The development of human metastatic cancer is a multistep process, involving the acquisition of several genetic mutations, tumour heterogeneity, and interactions with the surrounding microenvironment. Due to the complexity of cancer development in mammals, simpler model organisms, such as the vinegar fly, Drosophila melanogaster, are being utilized to provide novel insights into the molecular mechanisms involved. In this review, we highlight recent advances in modelling tumorigenesis using the Drosophila model, focusing on the cooperation of oncogenes or tumour suppressors, and the interaction of mutant cells with the surrounding tissue in epithelial tumour initiation and progression.
Figures

Cell competition mechanisms. The three main types of cell competition are shown. Mutant cells are in pink, wild-type cells are in blue, hemocytes are in grey, and the basement membrane (basal lamina) is in purple. (a) Classical cell competition: within an epithelium, cells with reduced levels of dMyc, ribosomal subunits mutants (minutes), Jak-Stat or Wg signalling, or high levels of Hippo signalling (losers) are eliminated by apoptosis, induced by the surrounding wild-type cells (winners). The loser cells express on their cell surface the Flower-Lose (FweLose) isoform (red dots), which marks them for elimination when in contact with the surrounding wild-type cells that express the Flower-Ubi (FweUbi) isoform (green dots). Additionally, signalling via the Spätzle ligand and Toll-Like Receptors (TLRs) in the loser cells triggers cell death via upregulation of cell death inducers, Rpr or Hid. Cells with upregulated Hippo signalling (or yki mutants) exhibit decreased dMyc levels, but cells with decreased ribosomal function, Jak-Stat, or Wg signalling undergo dMyc-independent cell competition. (b) Supercompetition: cells with high levels of dMyc, Jak-Stat, increased Wg signalling, or decreased Hippo signalling show “supercompetitor” behaviour and induce apoptosis in neighbouring wild-type cells. This occurs via the Flower-code or via Spätzle-TLR signalling in the loser cells. (c) Cell polarity mutant cell competition: cell polarity-impaired mutant cells are recognized by their epithelial neighbours or hemocytes (grey) and the TNFR-JNK signalling ligand, Egr (TNF), which is secreted by the wild-type epithelial cells or hemocytes. Mutant cells are removed by JNK-dependent and caspase-dependent apoptosis. JNK activation in neighbouring wild-type cells together with PVR, ELMO, and Mbc signalling is required in the wild-type cells for the removal of the dying cells. Hemocytes play the predominant role in engulfment and removal of the dead cells. The interaction of PTP10D in the mutant cell with SAS in the wild-type cell is important for “loser” cell fate of the polarity-impaired mutant cell. The Slit-Robo-Ena signalling pathway plays an important role in basal extrusion of the mutant cell, where the hemocytes are localized.
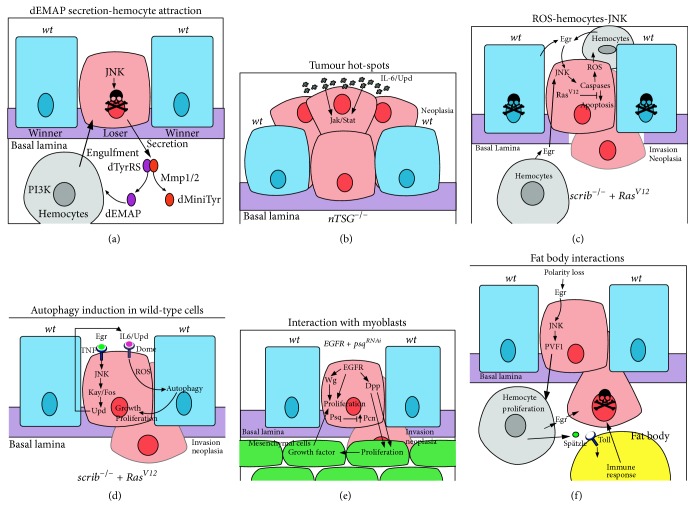
Cooperative interactions between the tumour and surrounding cells in tumorigenesis. Interactions between cells are shown that result in either the death of the mutant cell or cell survival, proliferation, and neoplastic transformation. Mutant cells are in pink, wild-type cells are in blue, hemocytes are in grey, myoblasts (mesenchymal cells) are in green, a fat body adipocyte is in yellow, and the basement membrane (basal lamina) is in purple. (a) dEMAP secretion-hemocyte attraction: JNK signalling in a cell polarity-impaired loser cell transcriptionally upregulates MMP1, which acts to cleave secreted dTyrRS to form dEMAP and dminiTyr. dEMAP attracts hemocytes to the loser cell by upregulating PI3K signalling in the hemocytes, which is required for chemotaxis and possibly engulfment of the loser cell. (b) Tumour hot-spots: neoplastic tumour-suppressor mutants (nTSGs) induce tumours more preferably, in regions where there is a stiff basal lamina and there are developmentally high levels of the Upd (IL-6) ligand to elevate Jak-Stat signalling, which promotes cell survival and proliferation of the tumour cells. (c) ROS-hemocytes-JNK: in scrib mutant RasV12-expressing tumour cells, a feedback loop between the hemocytes and the mutant cells promotes tumorigenesis. In the mutant cells, Ras signalling and caspase activation leads to ROS production that is released from the cells and promotes hemocytes to produce Egr (TNF). Egr signals via the TNFR-JNK pathway in the mutant cell leading to the upregulation of caspase activity, and some apoptosis, which is required for tumour overgrowth and invasion. Due to the disruption of the peripodial epithelium in large scrib mutant RasV12-expressing tumours, hemocytes most likely interact with the tumour on both apical and basal sides. (d) Induction of autophagy in surrounding wild-type cells: scrib mutant RasV12-expressing tumour cells are metabolically stressed, which leads to ROS production. Egr-JNK signalling leads to the transcriptional upregulation of Upd, ligands for the Dome-Jak-Stat signalling pathway, which is elevated in the mutant cells. Jak-Stat signalling and ROS production are required for the induction of autophagy in the surrounding wild-type cells, and also at distant sites, such as the fat body, muscle, and gut (not shown), which facilitates tumour growth and neoplastic transformation, possibly through supplying amino acids, glucose, and other nutrients to the tumour cells. (e) Interactions with myoblasts: in EGFR-overexpressing psq-knockdown tumours cooperative interactions are observed between the tumour cells and the surrounding myoblasts (mesenchymal cells). EGFR induces Wg and Dpp expression, and psq knockdown leads to increased levels of the extracellular matrix protein, Perlecan (Pcn). Wg acts to promote proliferation of the tumour cells, whilst Dpp, facilitated by Pcn in the basal lamina, stimulates proliferation of the myoblast cells. In turn, the myoblast cells provide unidentified growth factors that drive proliferation and neoplastic transformation of the tumour cells. Myoblasts also supply Egr (not shown), which would be expected to activate the TNFR-JNK signalling pathway in the tumour cells. (f) Interactions with the fat body: polarity-impaired tumours through Egr-JNK signalling upregulate PVF1, a ligand for the PVR receptor on hemocytes, which promotes hemocyte proliferation. Hemocytes, in turn, supply Egr to the tumour cells, and the Toll Receptor ligand, Spätzle, to the fat body, which induces innate immune system signalling in the fat body. These interactions are required to induce apoptosis of tumour cells.
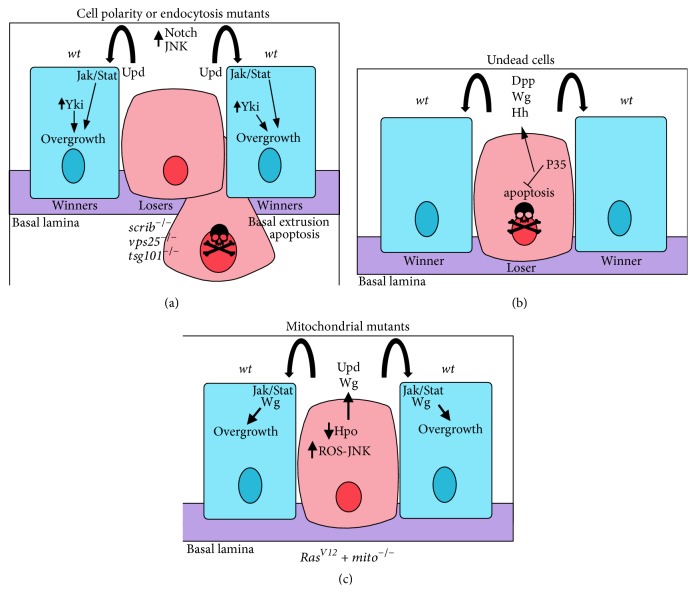
Non-cell-autonomous overgrowth. Examples of different types of non-cell-autonomous overgrowth. Mutant cells are in pink, wild-type cells are in blue, hemocytes are in grey, and the basement membrane (basal lamina) is in purple. (a) Cell polarity or endocytosis mutant cells are induced by JNK signalling to undergo cell death and induce non-cell-autonomous overgrowth of the surrounding wild-type cells. In vps25 or tsg101 (ept) endocytic mutants, which also show apicobasal cell polarity defects, ectopic activation of Notch signalling leads to the expression and secretion of the Dome-Jak-Stat pathway ligand, Upd, which promotes non-cell-autonomous proliferation and overgrowth of surrounding tissue. In scrib mutant cells, elevated JNK signalling, and impaired Hippo signalling, leads to transcriptional upregulation of Upd, which activates Dom-Jak-Stat signalling in the surrounding wild-type cells, thereby inducing their proliferation. (b) Undead cells, where apoptosis is initiated, but effector caspase activity is blocked, emit morphogens (such as Wg, Dpp, and Hh) that promote proliferation of their wild-type epithelial neighbours, thereby leading to non-cell-autonomous overgrowth. (c) Mitochondrial mutants expressing RasV12 lead to non-cell-autonomous overgrowth. The mitochondrial impairment results in the production of ROS, which induces JNK activation, which, in turn, results in Hippo pathway impairment, leading to expression of the Yki targets, Upd and Wg. Upd elevates Jak/Stat signalling and Wg induces Wg pathway signalling in the surrounding wild-type cells to promote their overgrowth.
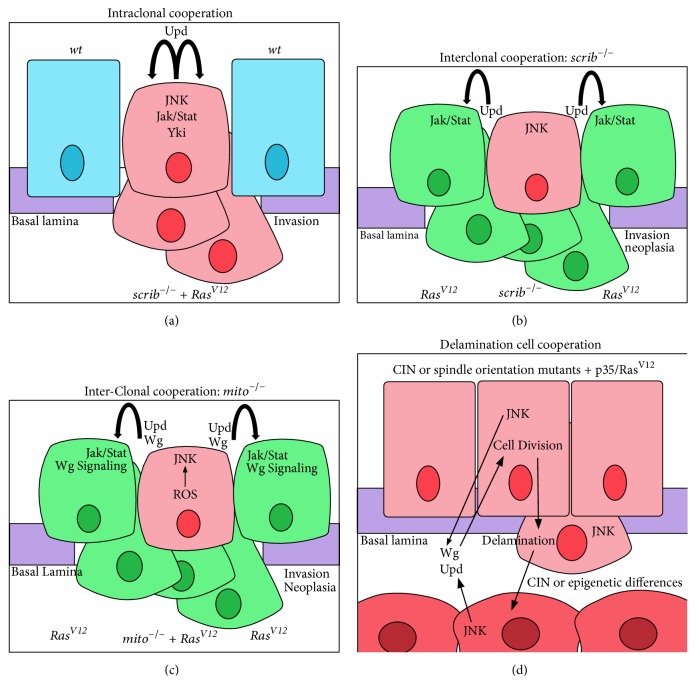
Different modes of cooperative tumorigenesis. Examples of different modes of cooperative tumorigenesis. Mutant cells are in pink, RasV12-expressing cells are in green, wild-type cells are in blue, delaminated mutant cells are in dark pink, and the basement membrane (basal lamina) is in purple. (a) Intraclonal cooperation with cell polarity mutants and RasV12: JNK activation in the tumour cells cooperates with oncogenic Ras signalling to promote tumour overgrowth and invasion. (b) Interclonal cooperation with cell polarity mutants and RasV12: JNK signalling and Hippo pathway impairment in the scrib mutant cells lead to the production of Upd, which induces Dome-Jak-Stat signalling in the surrounding RasV12-expressing cells, thereby inducing their overgrowth and invasion. (c) Interclonal cooperation with a mitochondrial mutant overexpressing RasV12 and RasV12-expressing surrounding cells: Upd and Wg are produced by the mitochondrial mutant RasV12-expressing surrounding cells (see Figure 3(c)), which induce upregulation of Dome-Jak-Stat and Wg signalling, respectively, in the RasV12 cells to induce their neoplastic overgrowth and invasion. (d) Delaminating cells cooperation: in tumours generated by chromosome instability (CIN) mutants (rod, bub3, and asp) or mutants that effect spindle orientation (scrib, dlg, and mud), some cells delaminate, resulting in two populations of cells, which in the case of spindle orientation mutants are not genetically different. The delaminated cell population produces the Wg and Upd ligands to upregulate Wg and Dome-Jak-Stat pathways, respectively, in the nondelaminated cells, thereby inducing their proliferation.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
