

Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics
- PMID: 29693319
- PMCID: PMC6002934
- DOI: 10.1002/wrna.1476
Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics
Abstract
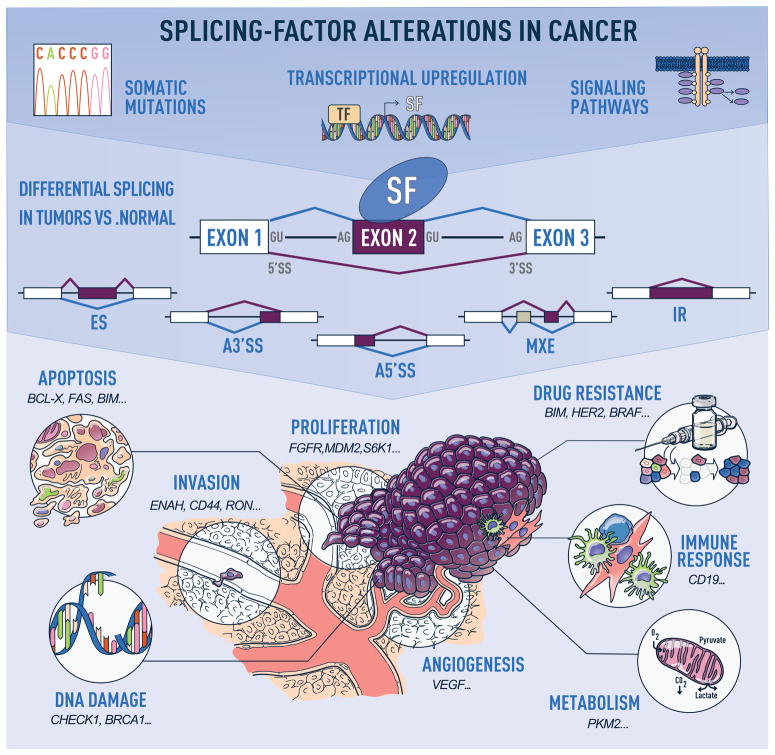
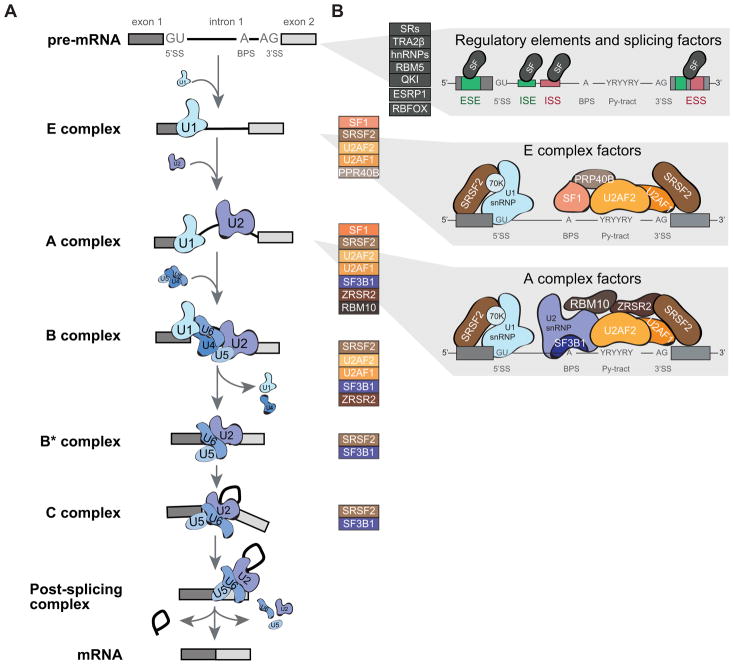
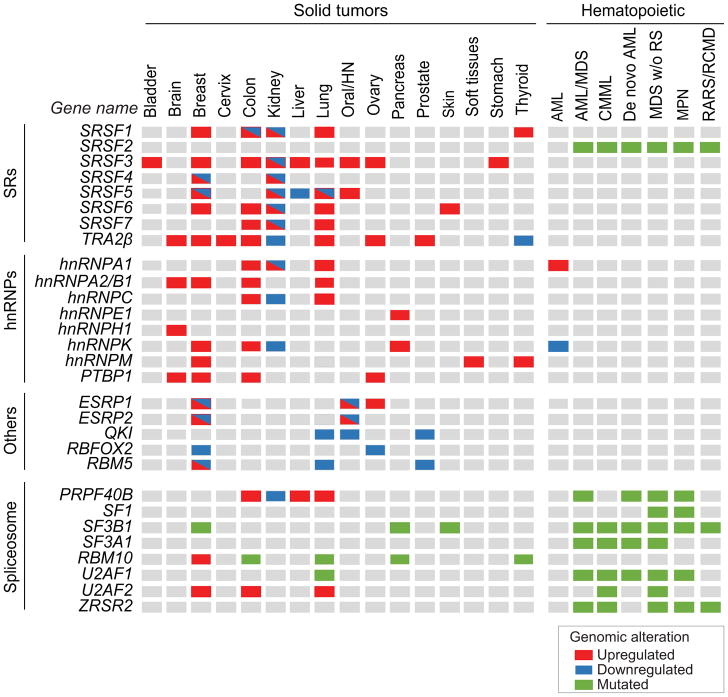
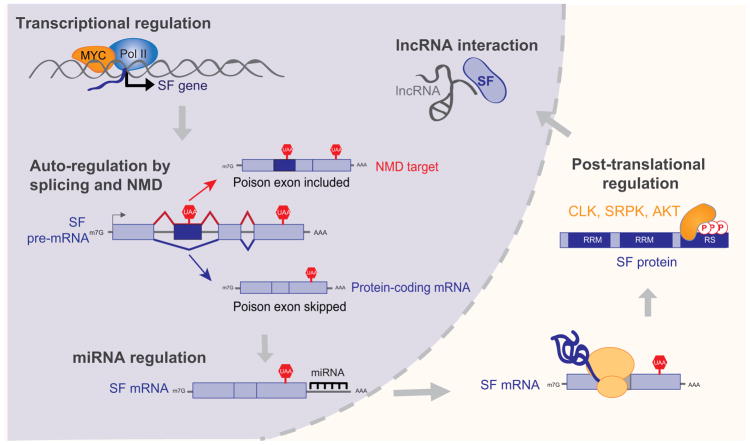
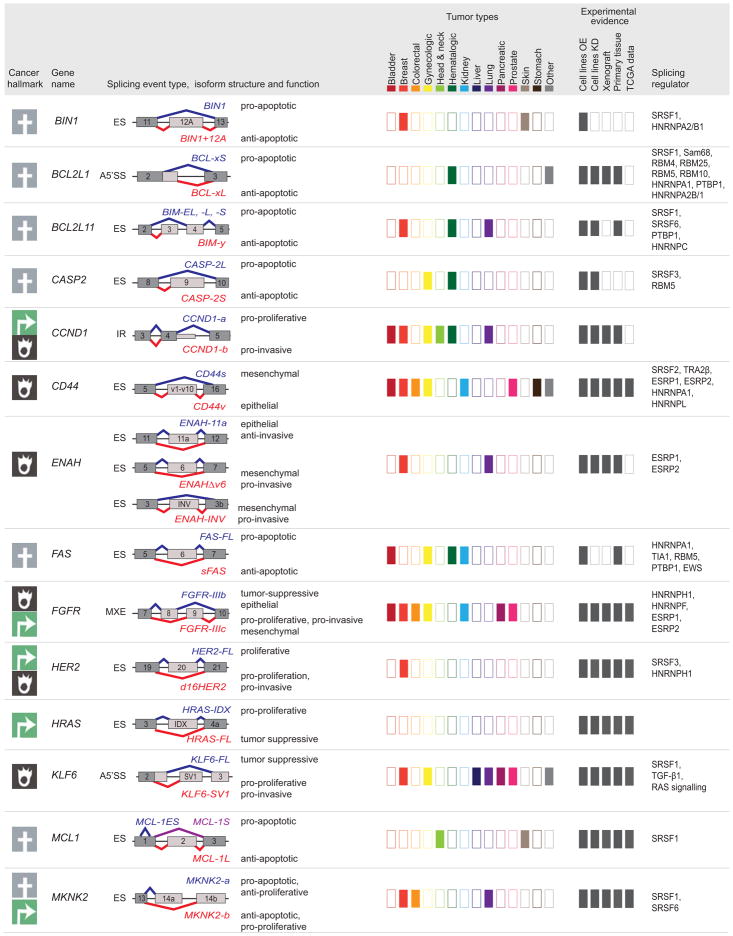
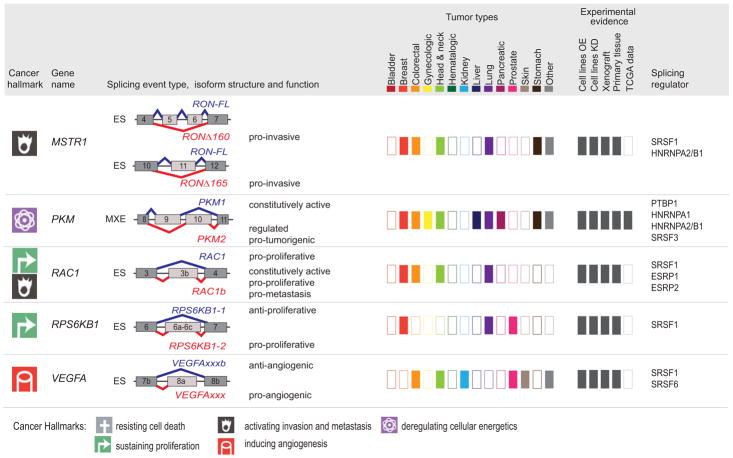
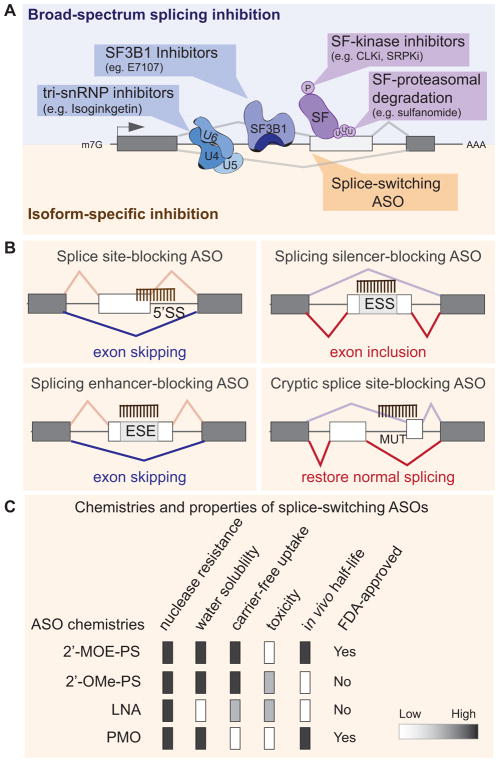
Defects in alternative splicing are frequently found in human tumors and result either from mutations in splicing-regulatory elements of specific cancer genes or from changes in the regulatory splicing machinery. RNA splicing regulators have emerged as a new class of oncoproteins and tumor suppressors, and contribute to disease progression by modulating RNA isoforms involved in the hallmark cancer pathways. Thus, dysregulation of alternative RNA splicing is fundamental to cancer and provides a potentially rich source of novel therapeutic targets. Here, we review the alterations in splicing regulatory factors detected in human tumors, as well as the resulting alternatively spliced isoforms that impact cancer hallmarks, and discuss how they contribute to disease pathogenesis. RNA splicing is a highly regulated process and, as such, the regulators are themselves tightly regulated. Differential transcriptional and posttranscriptional regulation of splicing factors modulates their levels and activities in tumor cells. Furthermore, the composition of the tumor microenvironment can also influence which isoforms are expressed in a given cell type and impact drug responses. Finally, we summarize current efforts in targeting alternative splicing, including global splicing inhibition using small molecules blocking the spliceosome or splicing-factor-modifying enzymes, as well as splice-switching RNA-based therapeutics to modulate cancer-specific splicing isoforms. This article is categorized under: RNA in Disease and Development > RNA in Disease RNA Processing > Splicing Regulation/Alternative Splicing.
Keywords: RNA binding proteins; RNA biology; RNA-based therapies; alternative splicing; antisense oligonucleotides; cancer; isoforms; oncogenes; spliceosome; splicing factors; tumor suppressors.
© 2018 Wiley Periodicals, Inc.
Figures
References
-
- Biamonti G, Catillo M, Pignataro D, Montecucco A, Ghigna C. The alternative splicing side of cancer. Semin Cell Dev Biol. 2014;32:30–36. - PubMed
-
- Bertram K, et al. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell. 2017;170(4):701–713 e711. - PubMed
-
- Hegele A, et al. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45(4):567–580. - PubMed
-
- Zhang X, Yan C, Hang J, Finci LI, Lei J, Shi Y. An Atomic Structure of the Human Spliceosome. Cell. 2017;169(5):918–929 e914. - PubMed
-
- Wahl MC, Luhrmann R. SnapShot: Spliceosome Dynamics I. Cell. 2015;161(6):1474–e1471. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
