

Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder
- PMID: 29693572
- PMCID: PMC6023449
- DOI: 10.3390/diagnostics8020029
Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder
Abstract
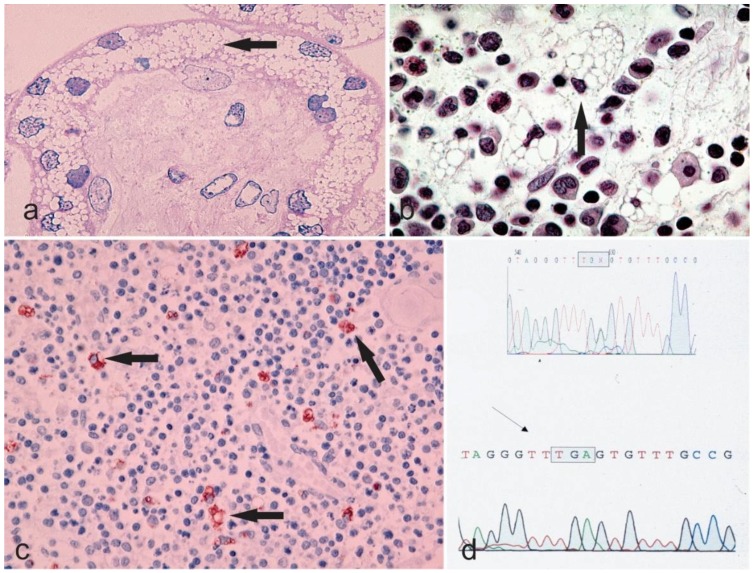
Sialidosis (MIM 256550) is a rare, autosomal recessive inherited disorder, caused by α-N-acetyl neuraminidase deficiency resulting from a mutation in the neuraminidase gene (NEU1), located on 6p21.33. This genetic alteration leads to abnormal intracellular accumulation as well as urinary excretion of sialyloligosaccharides. A definitive diagnosis is made after the identification of a mutation in the NEU1 gene. So far, 40 mutations of NEU1 have been reported. An association exists between the impact of the individual mutations and the severity of clinical presentation of sialidosis. According to the clinical symptoms, sialidosis has been divided into two subtypes with different ages of onset and severity, including sialidosis type I (normomorphic or mild form) and sialidosis type II (dysmorphic or severe form). Sialidosis II is further subdivided into (i) congenital; (ii) infantile; and (iii) juvenile. Despite being uncommon, sialidosis has enormous clinical relevance due to its debilitating character. A complete understanding of the underlying pathology remains a challenge, which in turn limits the development of effective therapeutic strategies. Furthermore, in the last few years, some atypical cases of sialidosis have been reported as well. We herein attempt to combine and discuss the underlying molecular biology, the clinical features, and the morphological patterns of sialidosis type I and II.
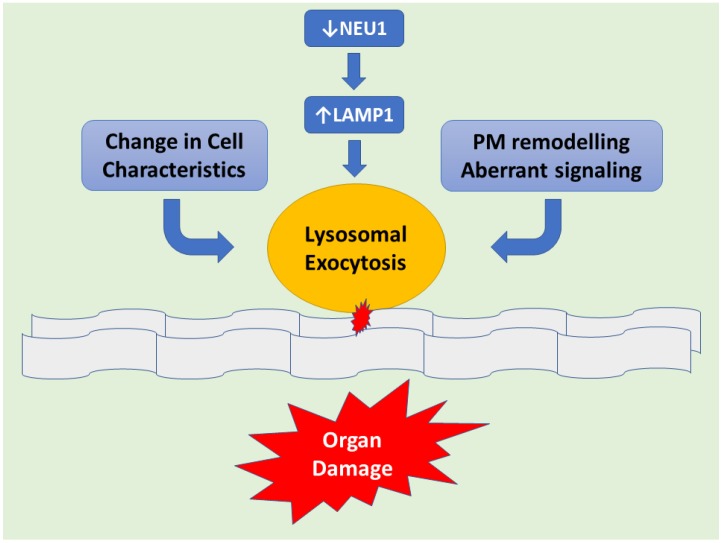
Keywords: lysosomal exocytosis; lysosomal storage disease; neuraminidase; sialidosis; sialidosis I; sialidosis II.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Pshezhetsky A.V., Richard C., Michaud L., Igdoura S., Wang S., Elsliger M.A., Qu J., Leclerc D., Gravel R., Dallaire L., et al. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nat. Genet. 1997;15:316–320. doi: 10.1038/ng0397-316. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
