

Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response
- PMID: 29693583
- PMCID: PMC6027212
- DOI: 10.3390/pathogens7020048
Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response
Abstract
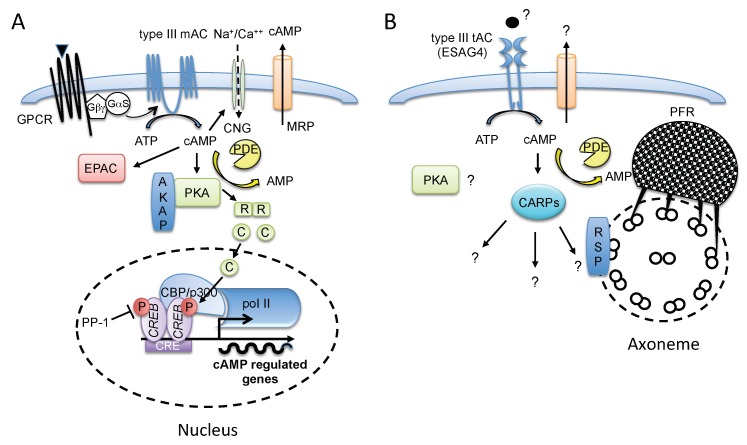
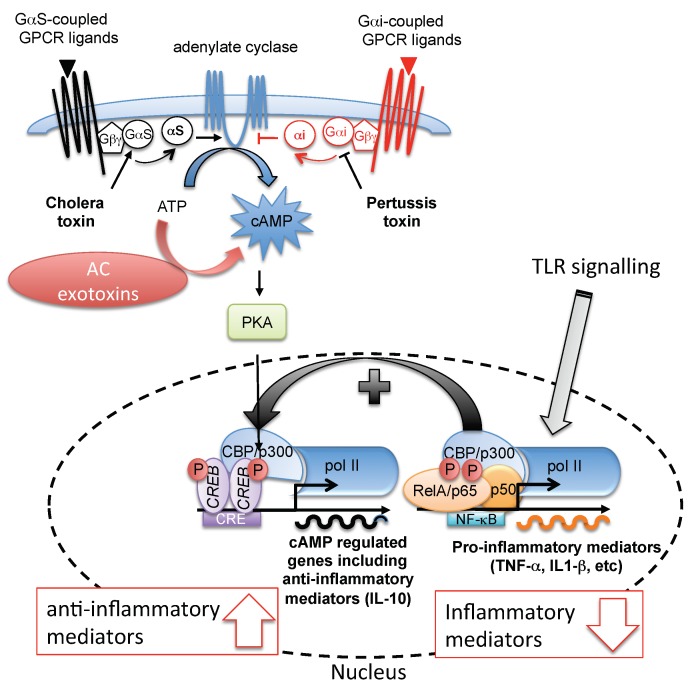
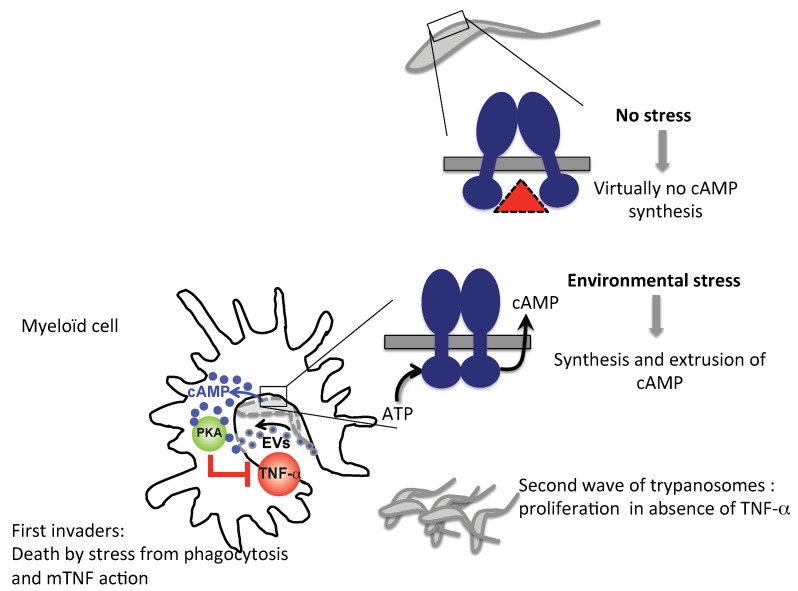
Trypanosoma brucei, etiological agent of Sleeping Sickness in Africa, is the prototype of African trypanosomes, protozoan extracellular flagellate parasites transmitted by saliva (Salivaria). In these parasites the molecular controls of the cell cycle and environmental sensing are elaborate and concentrated at the flagellum. Genomic analyses suggest that these parasites appear to differ considerably from the host in signaling mechanisms, with the exception of receptor-type adenylate cyclases (AC) that are topologically similar to receptor-type guanylate cyclase (GC) of higher eukaryotes but control a new class of cAMP targets of unknown function, the cAMP response proteins (CARPs), rather than the classical protein kinase A cAMP effector (PKA). T. brucei possesses a large polymorphic family of ACs, mainly associated with the flagellar membrane, and these are involved in inhibition of the innate immune response of the host prior to the massive release of immunomodulatory factors at the first peak of parasitemia. Recent evidence suggests that in T. brucei several insect-specific AC isoforms are involved in social motility, whereas only a few AC isoforms are involved in cytokinesis control of bloodstream forms, attesting that a complex signaling pathway is required for environmental sensing. In this review, after a general update on cAMP signaling pathway and the multiple roles of cAMP, I summarize the existing knowledge of the mechanisms by which pathogenic microorganisms modulate cAMP levels to escape immune defense.
Keywords: TNF-α; Trypanosoma brucei; adenylate cyclase; cAMP signaling; inflammation; innate immunity.
Conflict of interest statement
The author declares no conflict of interest.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
