

Integrative network analyses of wilt transcriptome in chickpea reveal genotype dependent regulatory hubs in immunity and susceptibility
- PMID: 29695764
- PMCID: PMC5916944
- DOI: 10.1038/s41598-018-19919-5
Integrative network analyses of wilt transcriptome in chickpea reveal genotype dependent regulatory hubs in immunity and susceptibility
Abstract
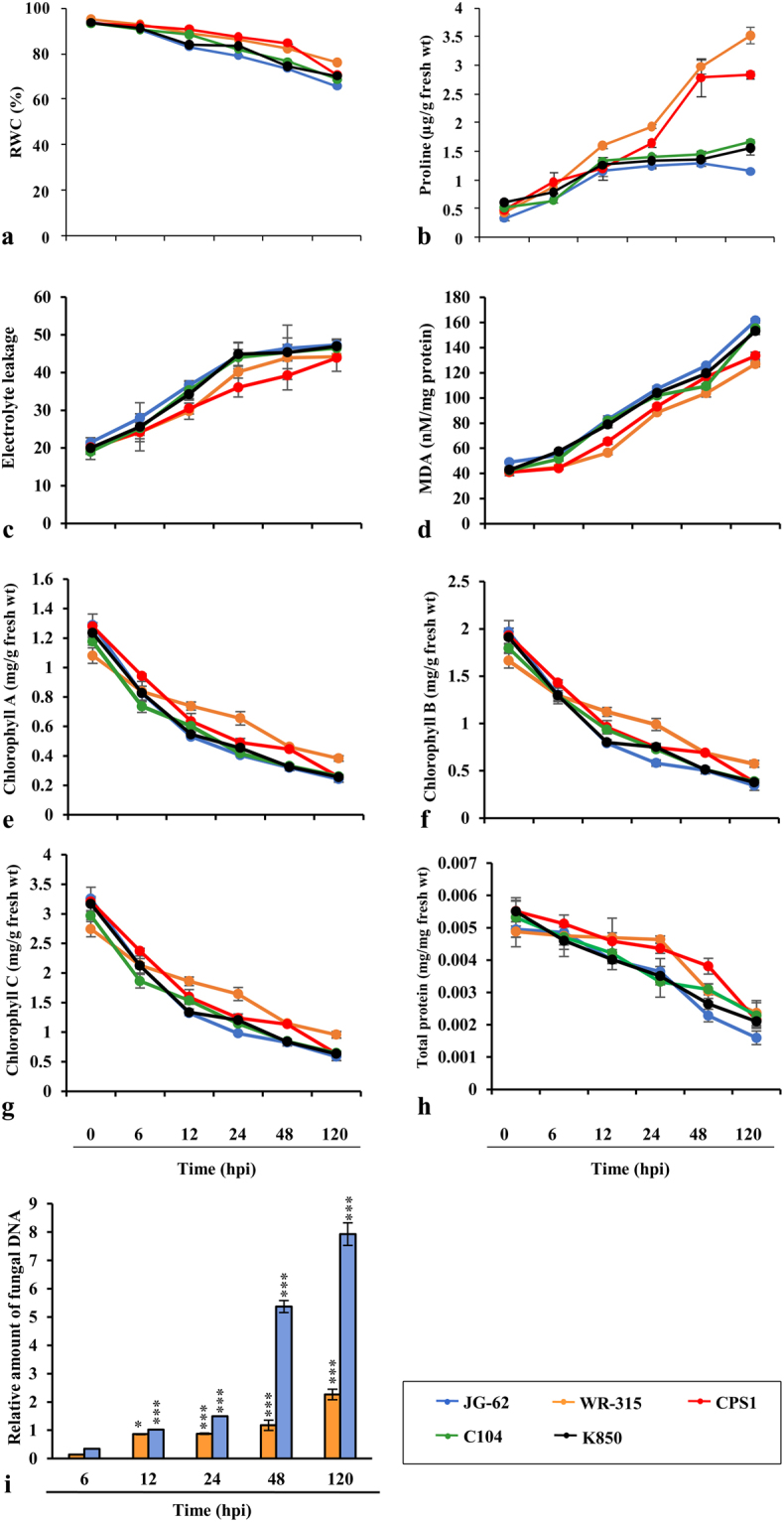
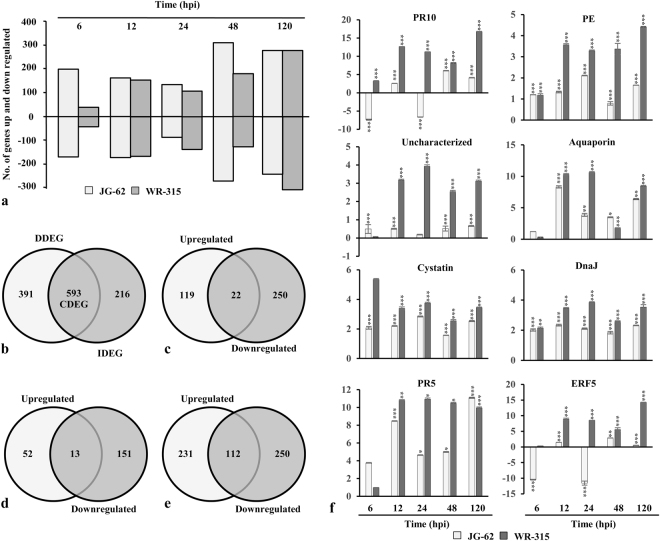
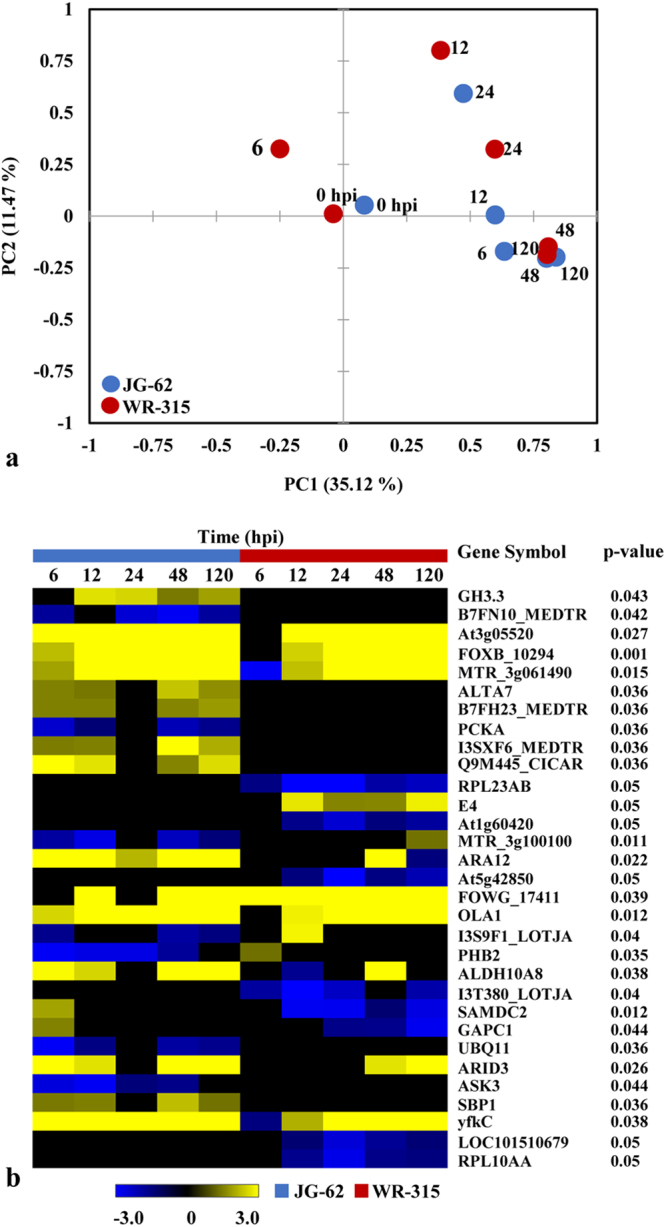
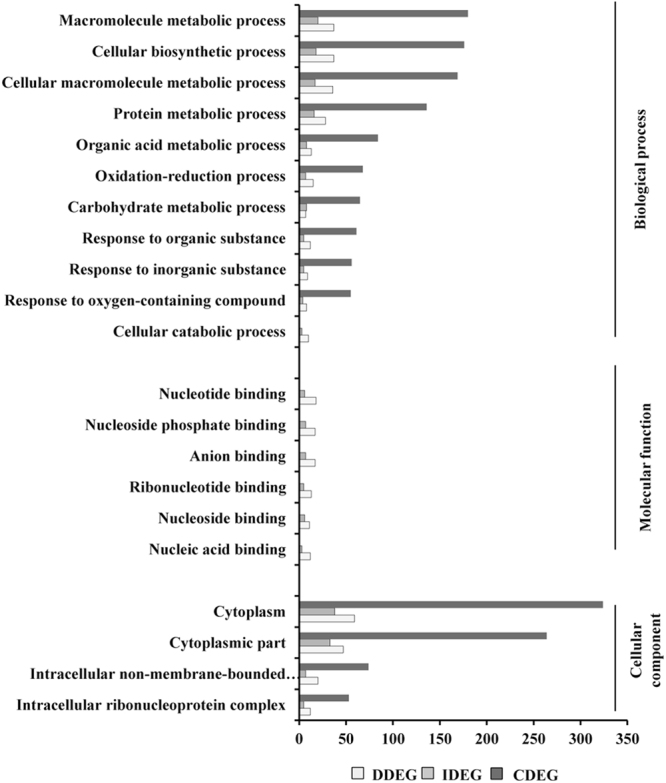
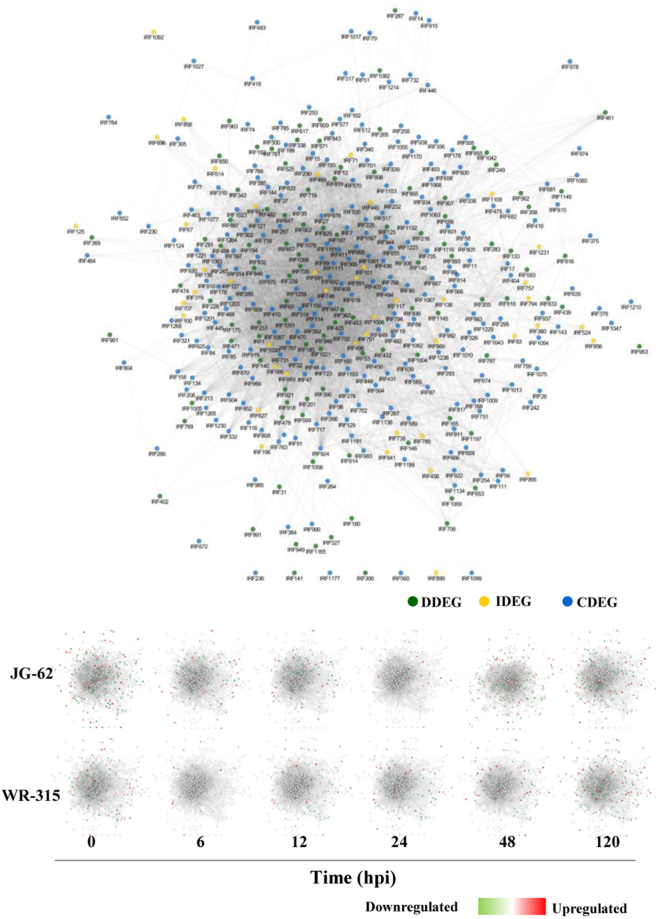
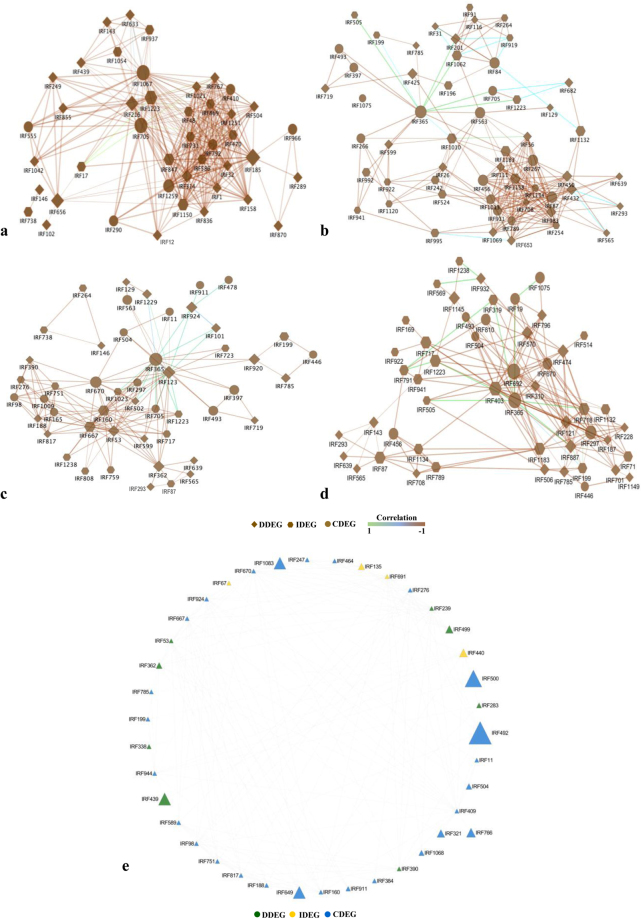
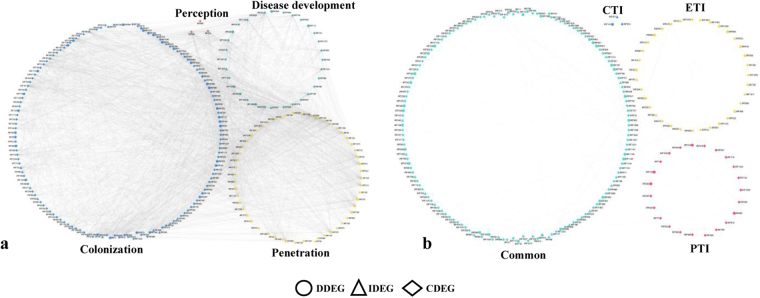
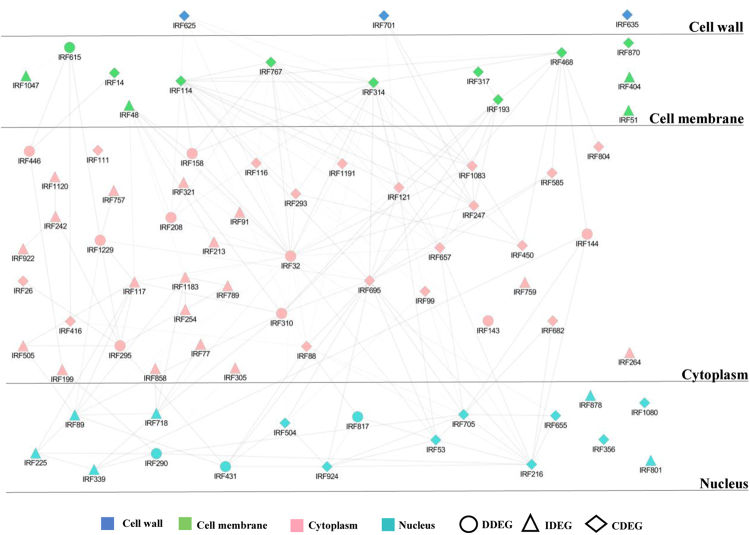
Host specific resistance and non-host resistance are two plant immune responses to counter pathogen invasion. Gene network organizing principles leading to quantitative differences in resistant and susceptible host during host specific resistance are poorly understood. Vascular wilt caused by root pathogen Fusarium species is complex and governed by host specific resistance in crop plants, including chickpea. Here, we temporally profiled two contrasting chickpea genotypes in disease and immune state to better understand gene expression switches in host specific resistance. Integrative gene-regulatory network elucidated tangible insight into interaction coordinators leading to pathway determination governing distinct (disease or immune) phenotypes. Global network analysis identified five major hubs with 389 co-regulated genes. Functional enrichment revealed immunome containing three subnetworks involving CTI, PTI and ETI and wilt diseasome encompassing four subnetworks highlighting pathogen perception, penetration, colonization and disease establishment. These subnetworks likely represent key components that coordinate various biological processes favouring defence or disease. Furthermore, we identified core 76 disease/immunity related genes through subcellular analysis. Our regularized network with robust statistical assessment captured known and unexpected gene interaction, candidate novel regulators as future biomarkers and first time showed system-wide quantitative architecture corresponding to genotypic characteristics in wilt landscape.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
