

Cutaneous neurofibromas in the genomics era: current understanding and open questions
- PMID: 29695767
- PMCID: PMC6008439
- DOI: 10.1038/s41416-018-0073-2
Cutaneous neurofibromas in the genomics era: current understanding and open questions
Abstract
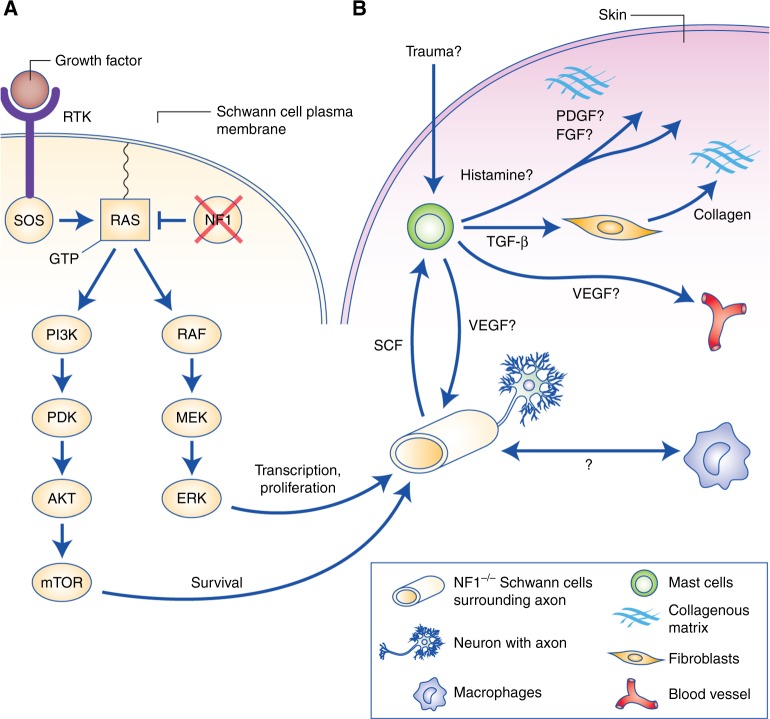
Cutaneous neurofibromas (cNF) are a nearly ubiquitous symptom of neurofibromatosis type 1 (NF1), a disorder with a broad phenotypic spectrum caused by germline mutation of the neurofibromatosis type 1 tumour suppressor gene (NF1). Symptoms of NF1 can include learning disabilities, bone abnormalities and predisposition to tumours such as cNFs, plexiform neurofibromas, malignant peripheral nerve sheath tumours and optic nerve tumours. There are no therapies currently approved for cNFs aside from elective surgery, and the molecular aetiology of cNF remains relatively uncharacterised. Furthermore, whereas the biallelic inactivation of NF1 in neoplastic Schwann cells is critical for cNF formation, it is still unclear which additional genetic, transcriptional, epigenetic, microenvironmental or endocrine changes are important. Significant inroads have been made into cNF understanding, including NF1 genotype-phenotype correlations in NF1 microdeletion patients, the identification of recurring somatic mutations, studies of cNF-invading mast cells and macrophages, and clinical trials of putative therapeutic targets such as mTOR, MEK and c-KIT. Despite these advances, several gaps remain in our knowledge of the associated pathogenesis, which is further hampered by a lack of translationally relevant animal models. Some of these questions may be addressed in part by the adoption of genomic analysis techniques. Understanding the aetiology of cNF at the genomic level may assist in the development of new therapies for cNF, and may also contribute to a greater understanding of NF1/RAS signalling in cancers beyond those associated with NF1. Here, we summarise the present understanding of cNF biology, including the pathogenesis, mutational landscape, contribution of the tumour microenvironment and endocrine signalling, and the historical and current state of clinical trials for cNF. We also highlight open access data resources and potential avenues for future research that leverage recently developed genomics-based methods in cancer research.
Conflict of interest statement
S.L.R., P.K. and A.B. are employees of The Children’s Tumor Foundation, which is a prominent non-profit funding organisation within the neurofibromatosis research community.
Figures

References
-
- Evans, D. G. et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am. J. Med. Genet. A152A, 327–332 (2010). - PubMed
-
- Lammert, M., Friedman, J. M., Kluwe, L. & Mautner, V. F. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch. Dermatol. 141, 71–74 (2005). - PubMed
-
- Friedman, J. Neurofibromatosis 1 [Internet]. Gene reviews. University of Washington. http://www.ncbi.nlm.nih.gov/pubmed/20301288. Accessed 6 July 2016 (2014).
-
- Recklinghausen, F. von. Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen [Internet]. https://wellcomelibrary.org. Accessed 8 August 2017 (1882).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
