

Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies
- PMID: 29695774
- PMCID: PMC5916929
- DOI: 10.1038/s41467-018-03766-z
Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies
Abstract
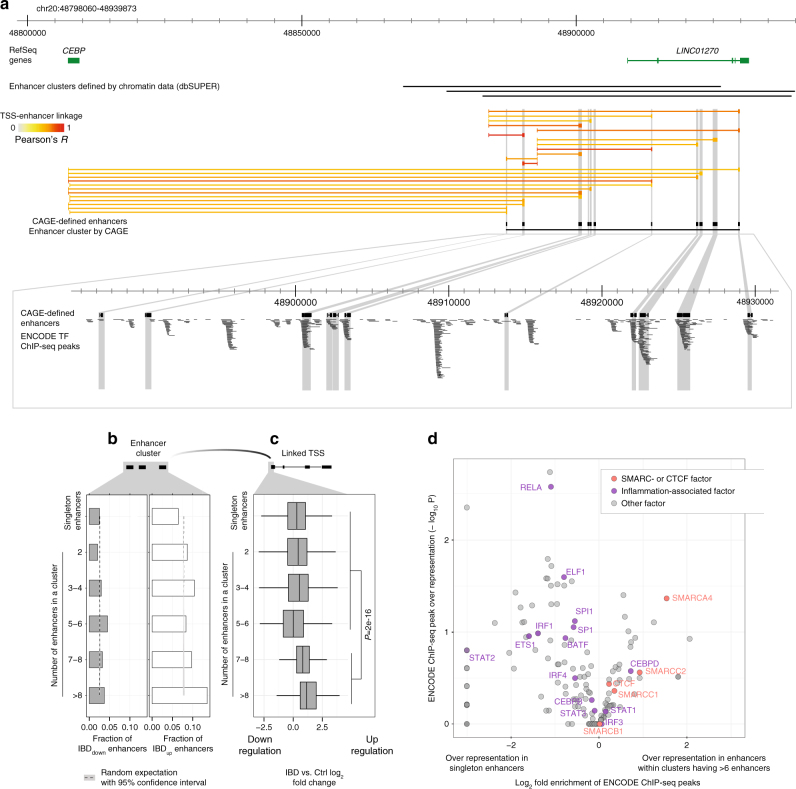
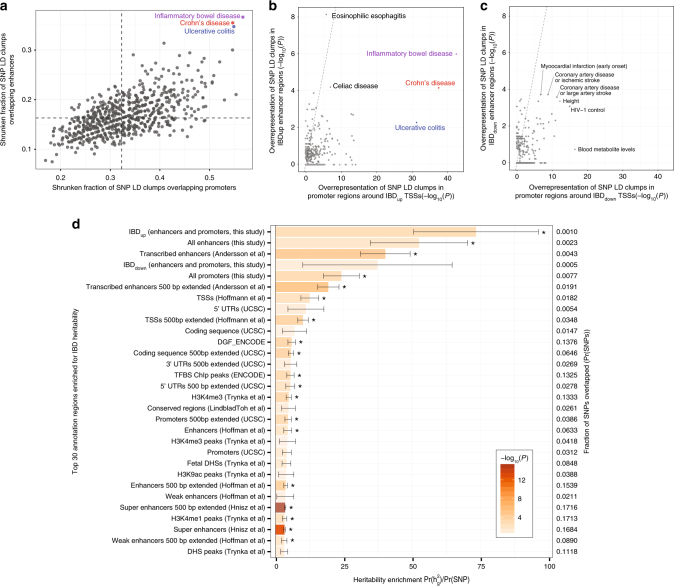
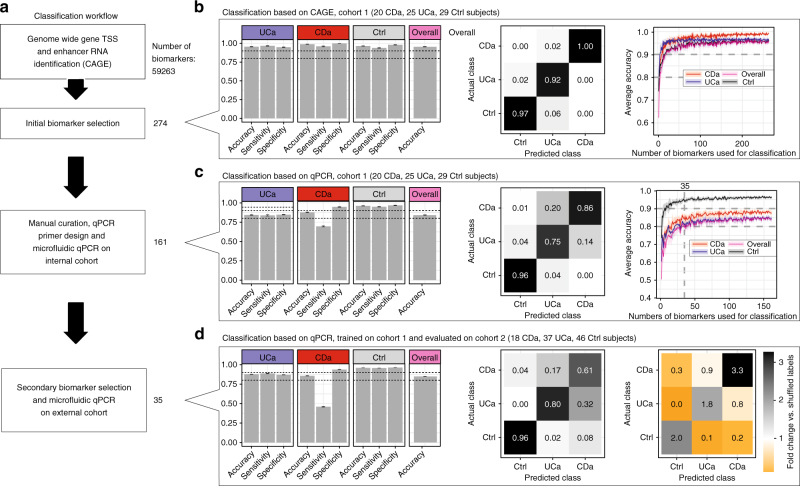
Inflammatory bowel disease (IBD) is a chronic intestinal disorder, with two main types: Crohn's disease (CD) and ulcerative colitis (UC), whose molecular pathology is not well understood. The majority of IBD-associated SNPs are located in non-coding regions and are hard to characterize since regulatory regions in IBD are not known. Here we profile transcription start sites (TSSs) and enhancers in the descending colon of 94 IBD patients and controls. IBD-upregulated promoters and enhancers are highly enriched for IBD-associated SNPs and are bound by the same transcription factors. IBD-specific TSSs are associated to genes with roles in both inflammatory cascades and gut epithelia while TSSs distinguishing UC and CD are associated to gut epithelia functions. We find that as few as 35 TSSs can distinguish active CD, UC, and controls with 85% accuracy in an independent cohort. Our data constitute a foundation for understanding the molecular pathology, gene regulation, and genetics of IBD.
Conflict of interest statement
The authors A.S., M.B., J.B., M.V., M.T., K.V.S., O.H.N. and J.B. have filed a patent for the method for classifying CD/UC vs. control based on qPCR, based on this study. The remaining authors declare no competing interests.
Figures
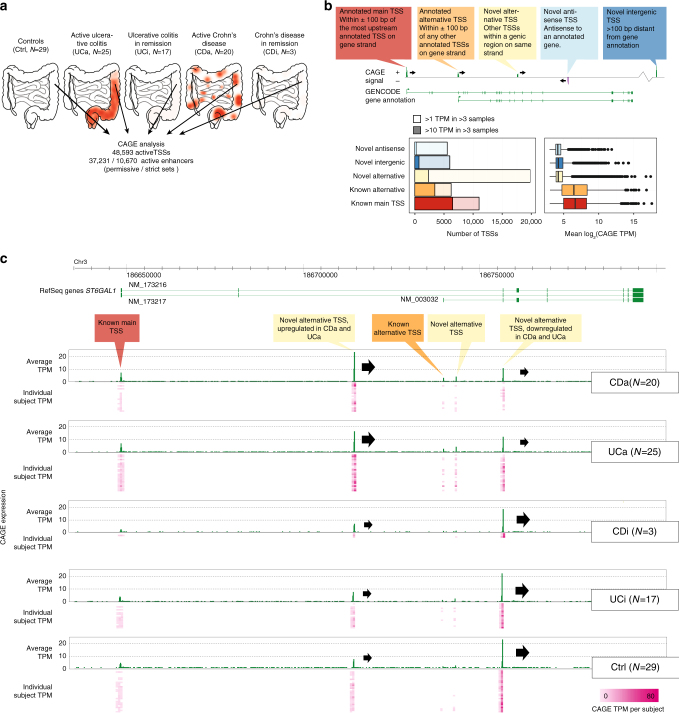
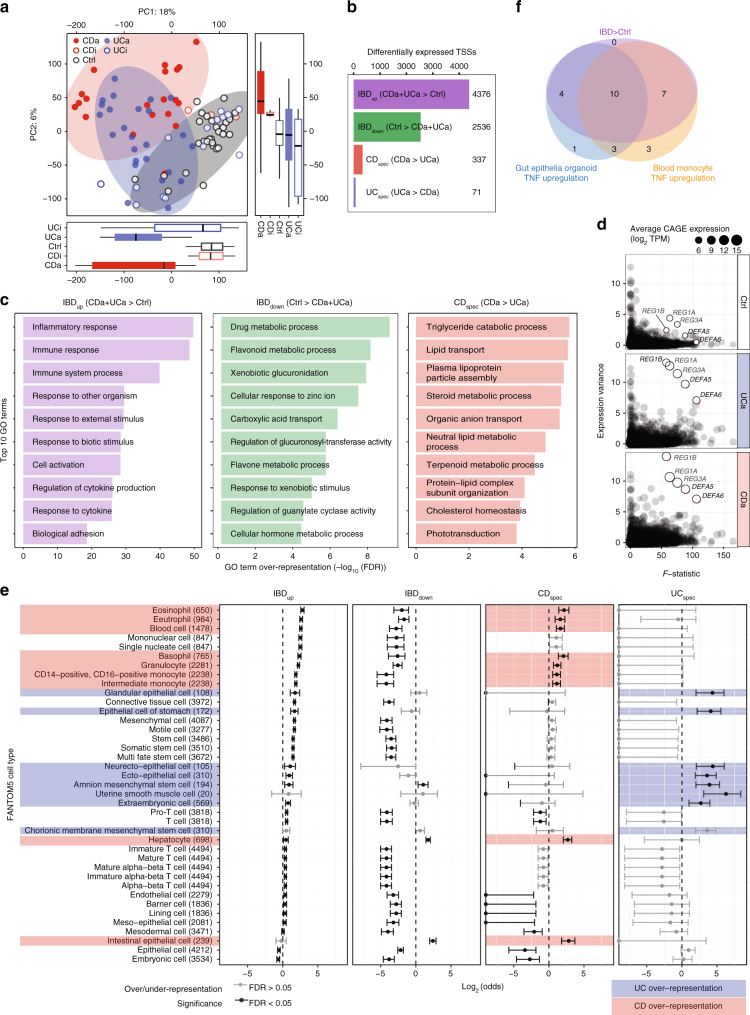
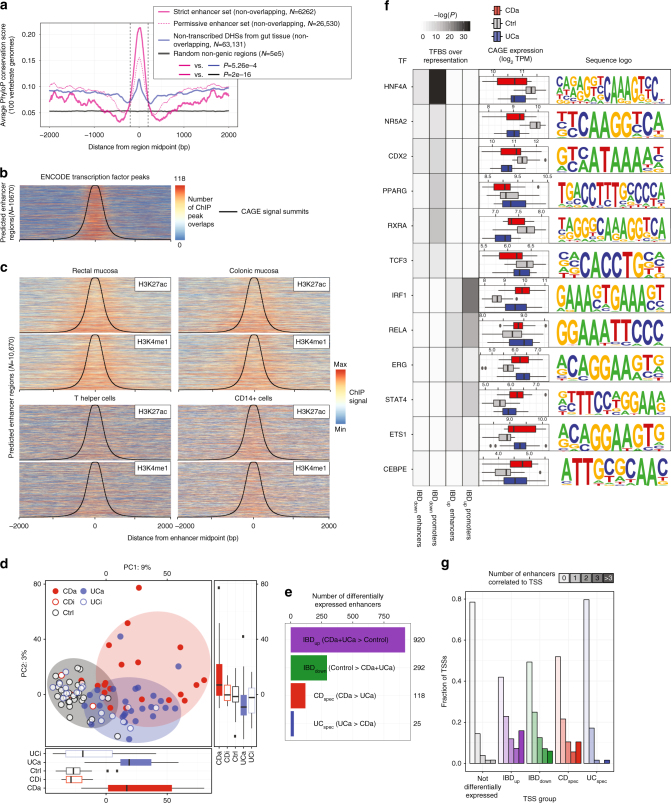
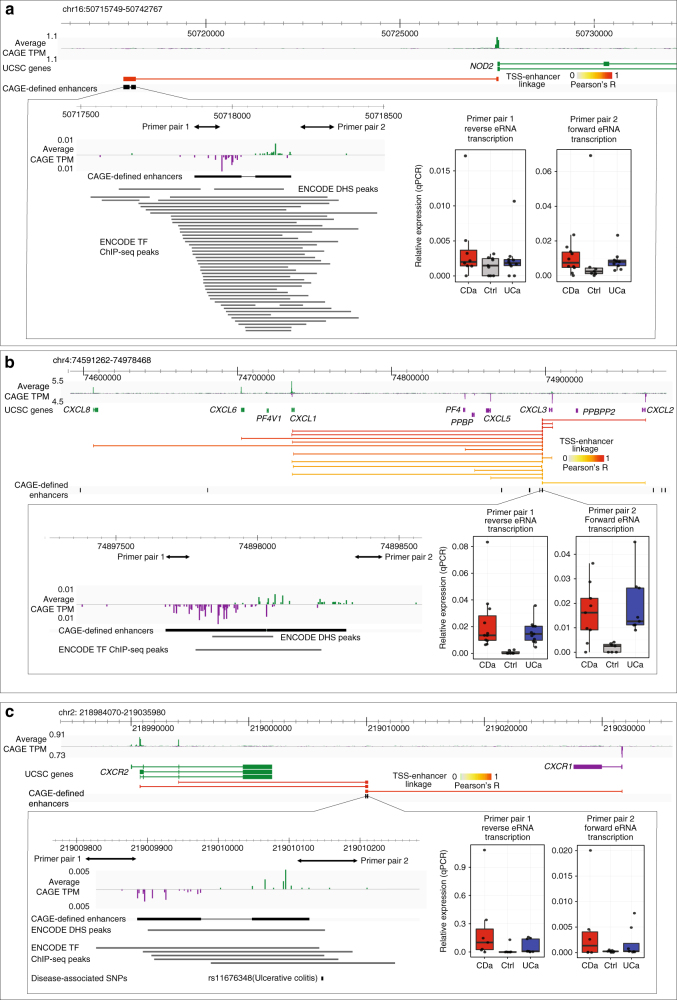
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
