

Genomic variation in 3,010 diverse accessions of Asian cultivated rice
- PMID: 29695866
- PMCID: PMC6784863
- DOI: 10.1038/s41586-018-0063-9
Genomic variation in 3,010 diverse accessions of Asian cultivated rice
Abstract
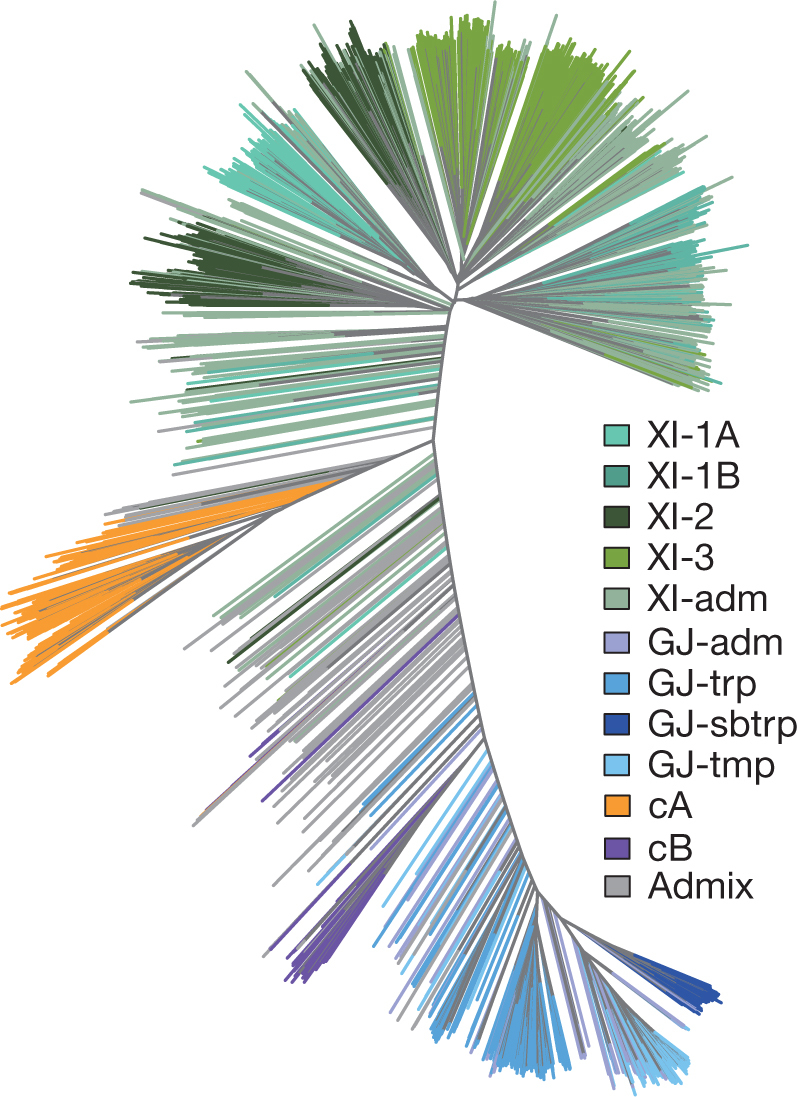
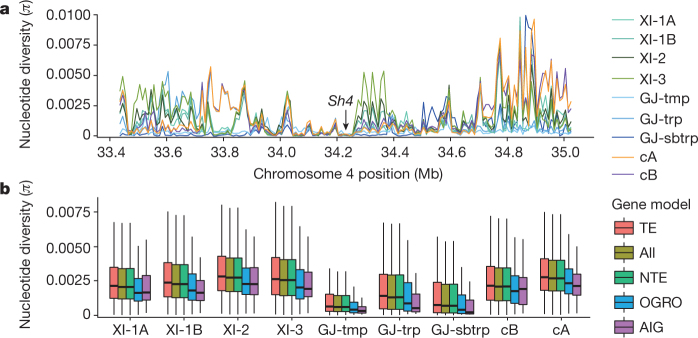
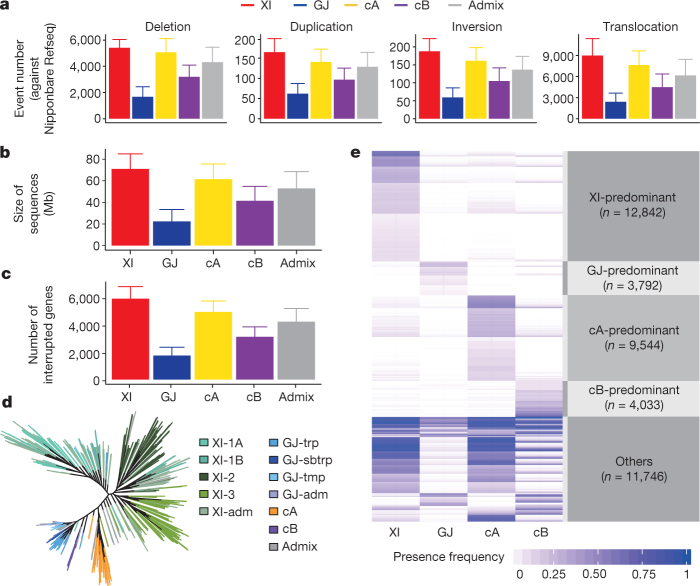
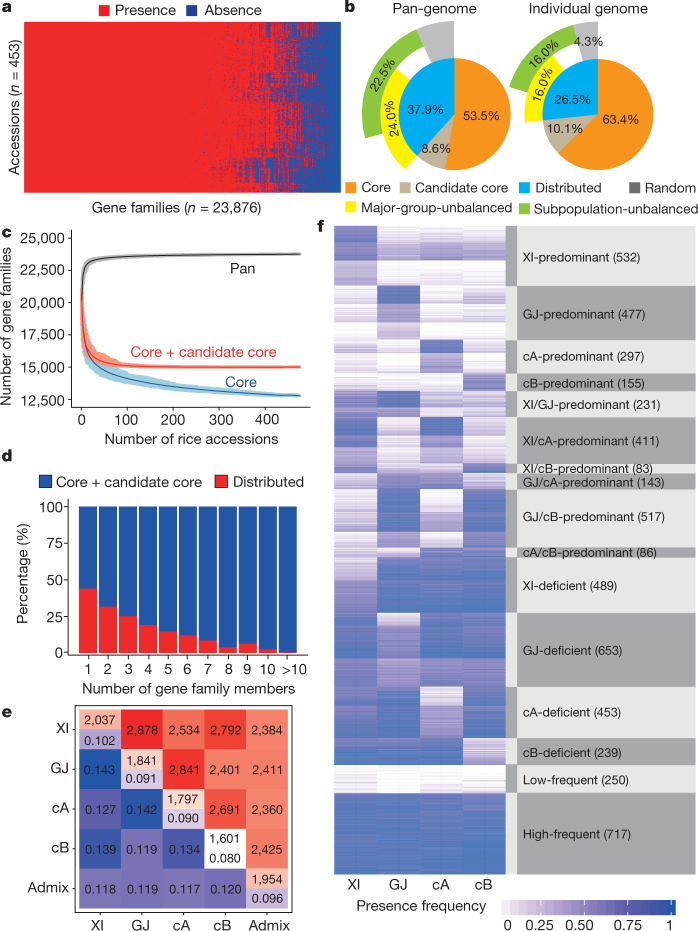
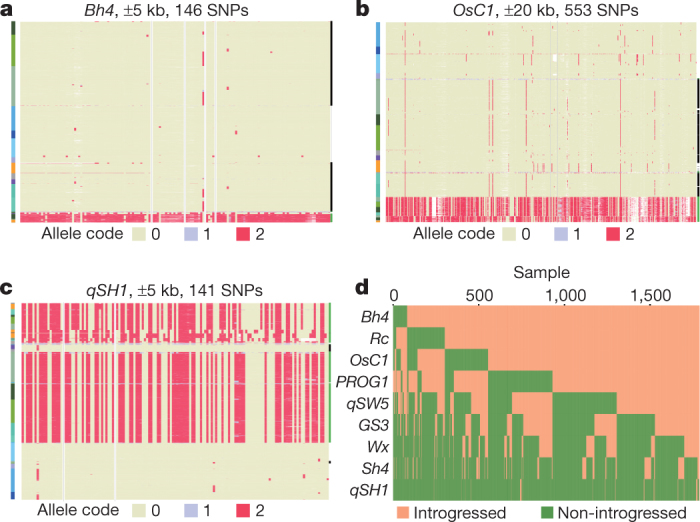
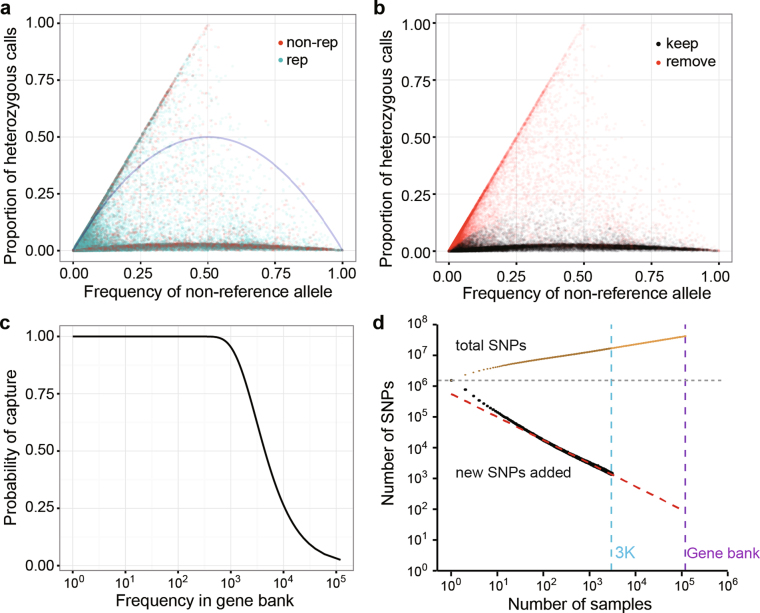
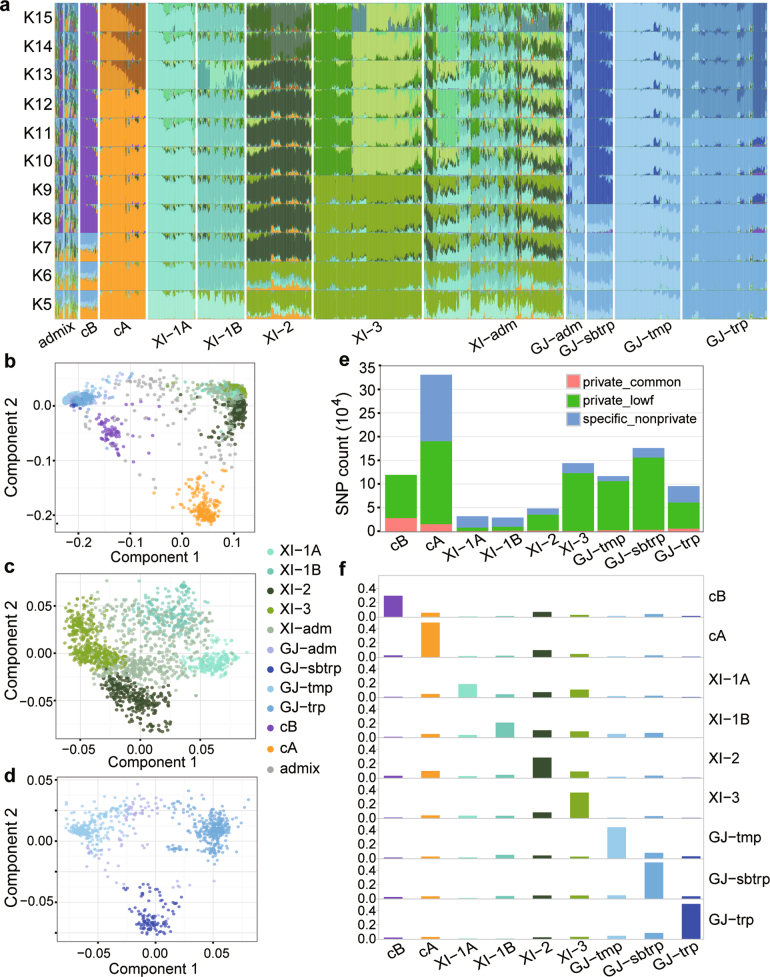
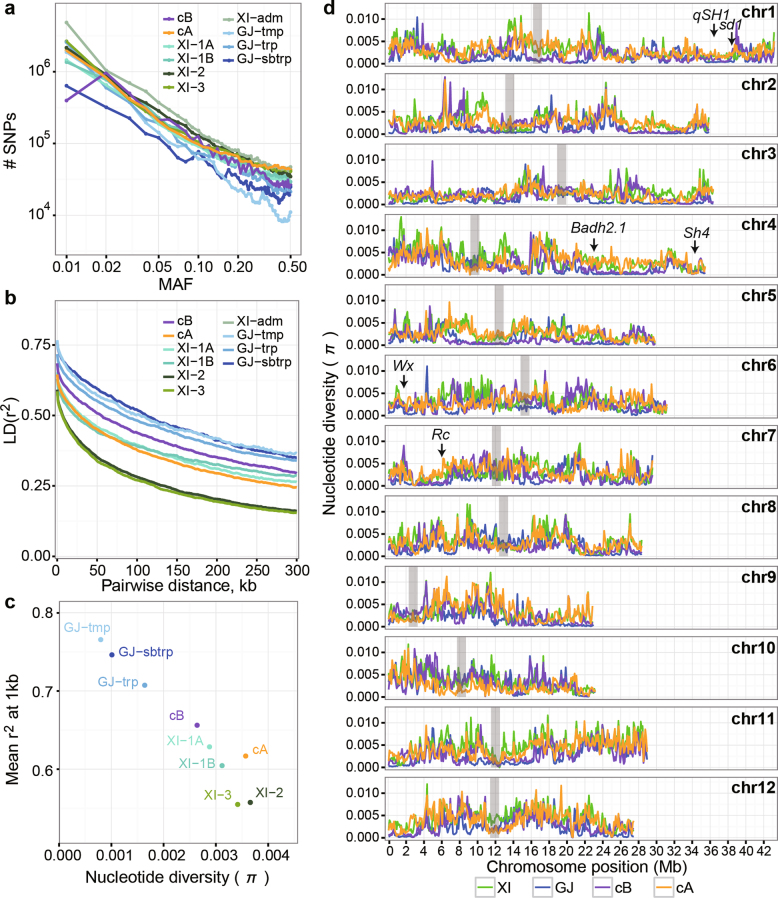
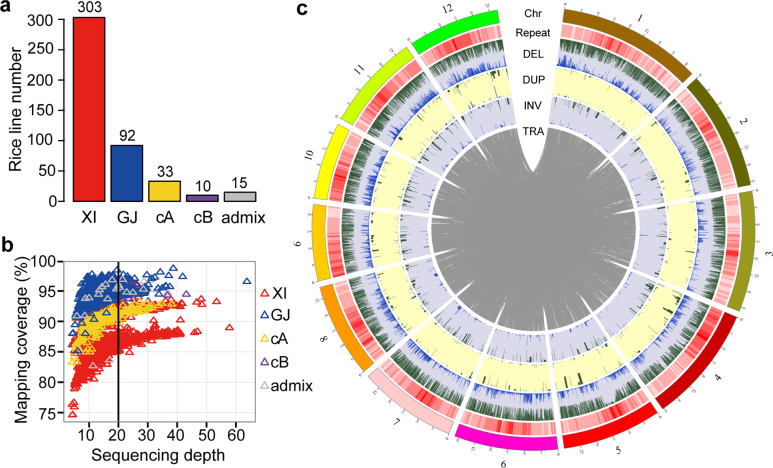
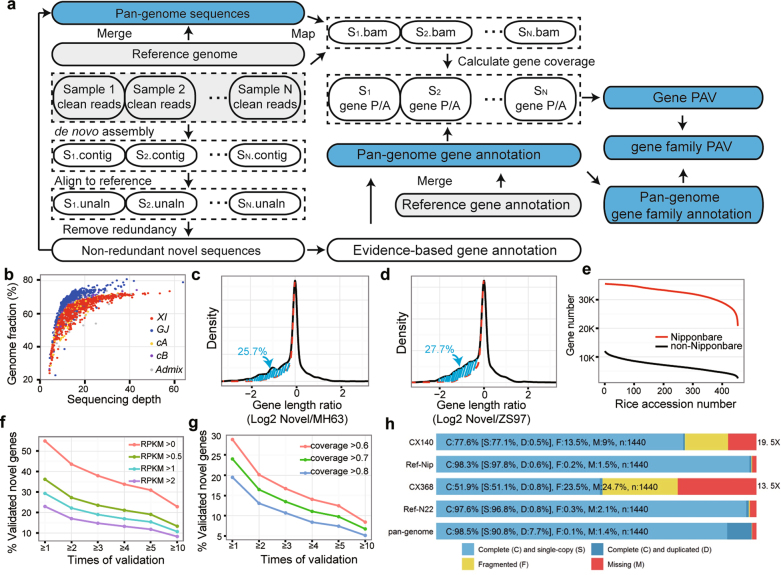
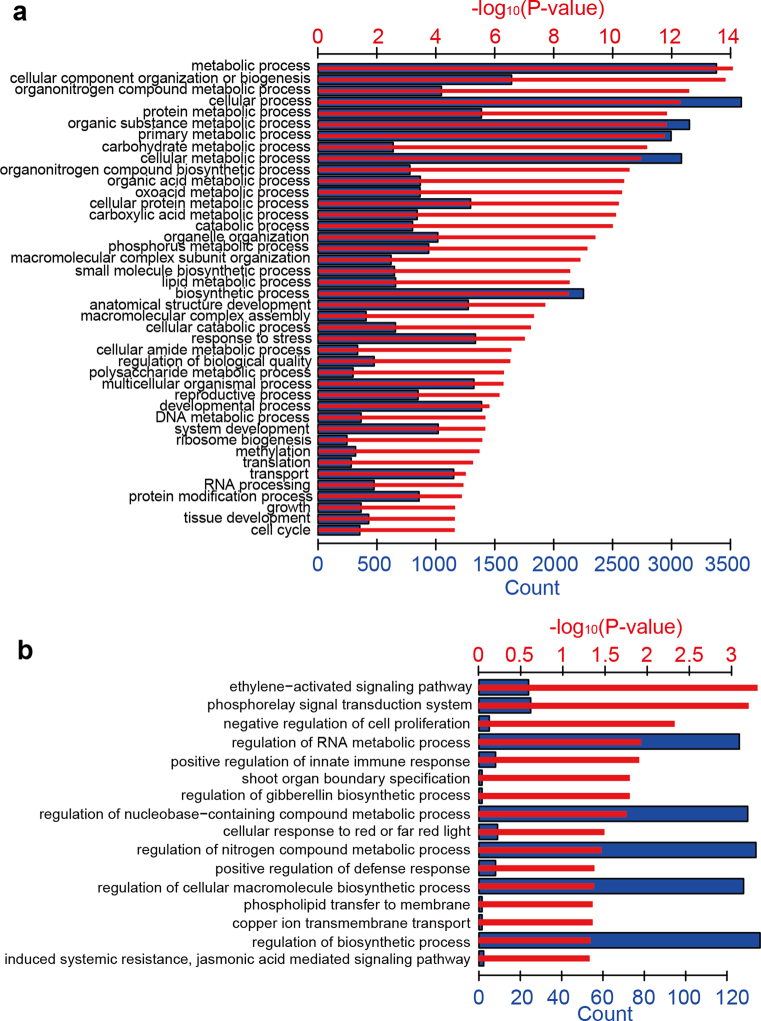
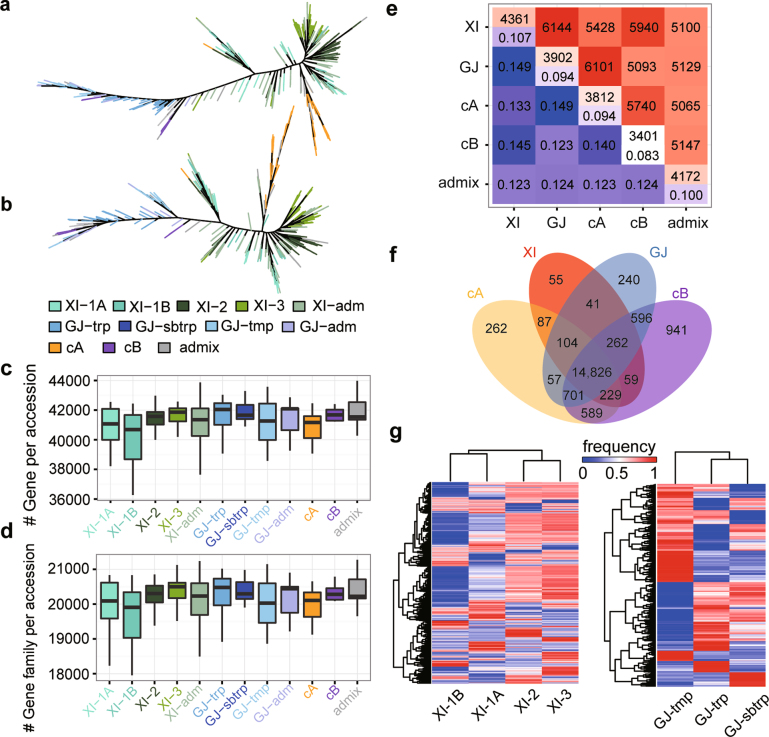
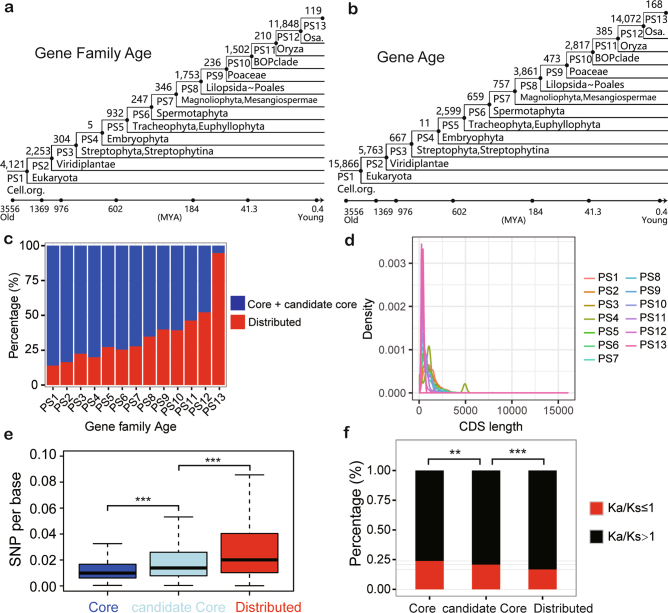
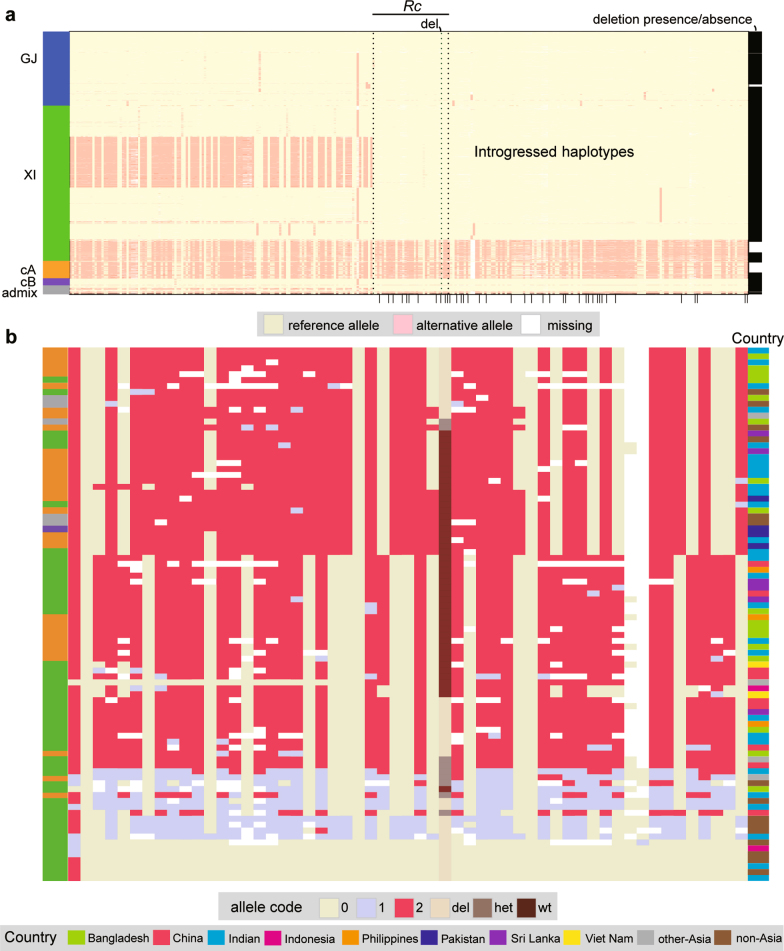
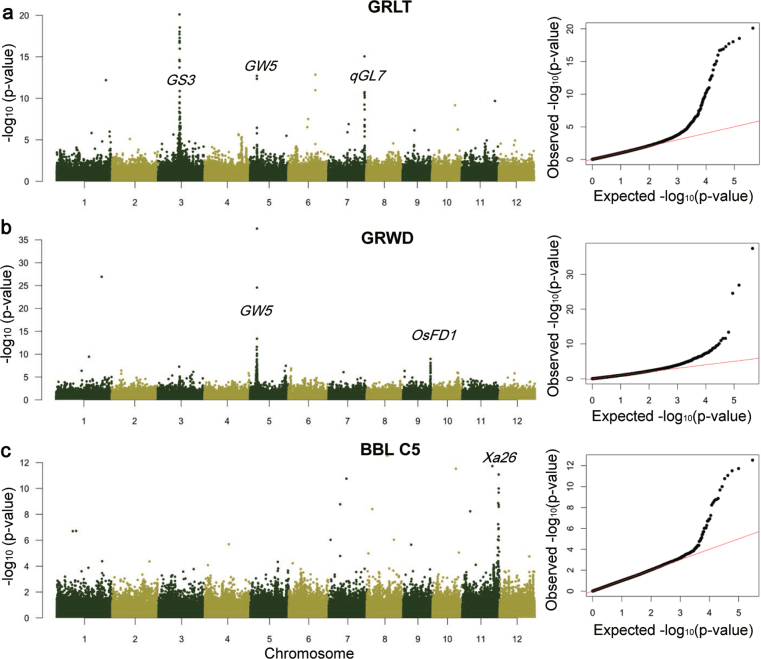
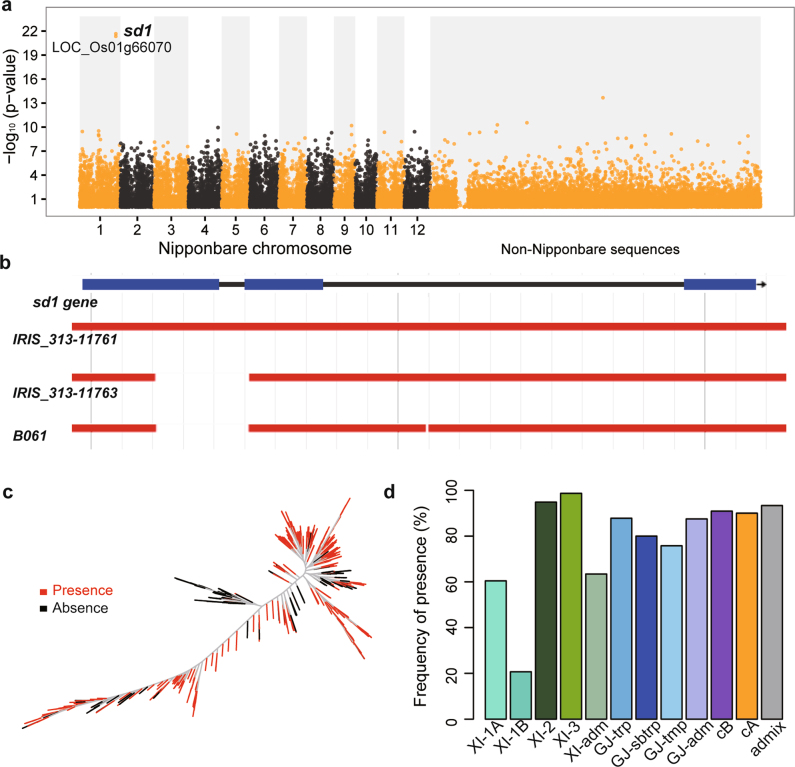
Here we analyse genetic variation, population structure and diversity among 3,010 diverse Asian cultivated rice (Oryza sativa L.) genomes from the 3,000 Rice Genomes Project. Our results are consistent with the five major groups previously recognized, but also suggest several unreported subpopulations that correlate with geographic location. We identified 29 million single nucleotide polymorphisms, 2.4 million small indels and over 90,000 structural variations that contribute to within- and between-population variation. Using pan-genome analyses, we identified more than 10,000 novel full-length protein-coding genes and a high number of presence-absence variations. The complex patterns of introgression observed in domestication genes are consistent with multiple independent rice domestication events. The public availability of data from the 3,000 Rice Genomes Project provides a resource for rice genomics research and breeding.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Seck PA, Diagne A, Mohanty S, Wopereis MC. Crops that feed the world 7: rice. Food Secur. 2012;4:7–24. doi: 10.1007/s12571-012-0168-1. - DOI
-
- Ting Y. Origination of the rice cultivation in China. J. College of Agric. Sun Yat-Sen University. 1949;7:11–24.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
