

Molecular regulations and therapeutic targets of Gaucher disease
- PMID: 29699937
- PMCID: PMC8108120
- DOI: 10.1016/j.cytogfr.2018.04.003
Molecular regulations and therapeutic targets of Gaucher disease
Abstract
Gaucher disease (GD) is the most common lysosomal storage disease caused by deficiency of beta-glucocerebrosidase (GCase) resulting in lysosomal accumulation of its glycolipid substrate glucosylceramide. The activity of GCase depends on many factors such as proper folding and lysosomal localization, which are influenced by mutations in GCase encoding gene, and regulated by various GCase-binding partners including Saposin C, progranulin and heat shock proteins. In addition, proinflammatory molecules also contribute to pathogenicity of GD. In this review, we summarize the molecules that are known to be important for the pathogenesis of GD, particularly those modulating GCase lysosomal appearance and activity. In addition, small molecules that inhibit inflammatory mediators, calcium ion channels and other factors associated with GD are also described. Discovery and characterization of novel molecules that impact GD are not only important for deciphering the pathogenic mechanisms of the disease, but they also provide new targets for drug development to treat the disease.
Keywords: Beta-glucocerebrosidase; Gaucher disease; Heat shock proteins; LIMP-2; Progranulin; Saposin C.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Conflict of interest
We herein declare that we have no conflict of interest.
Figures
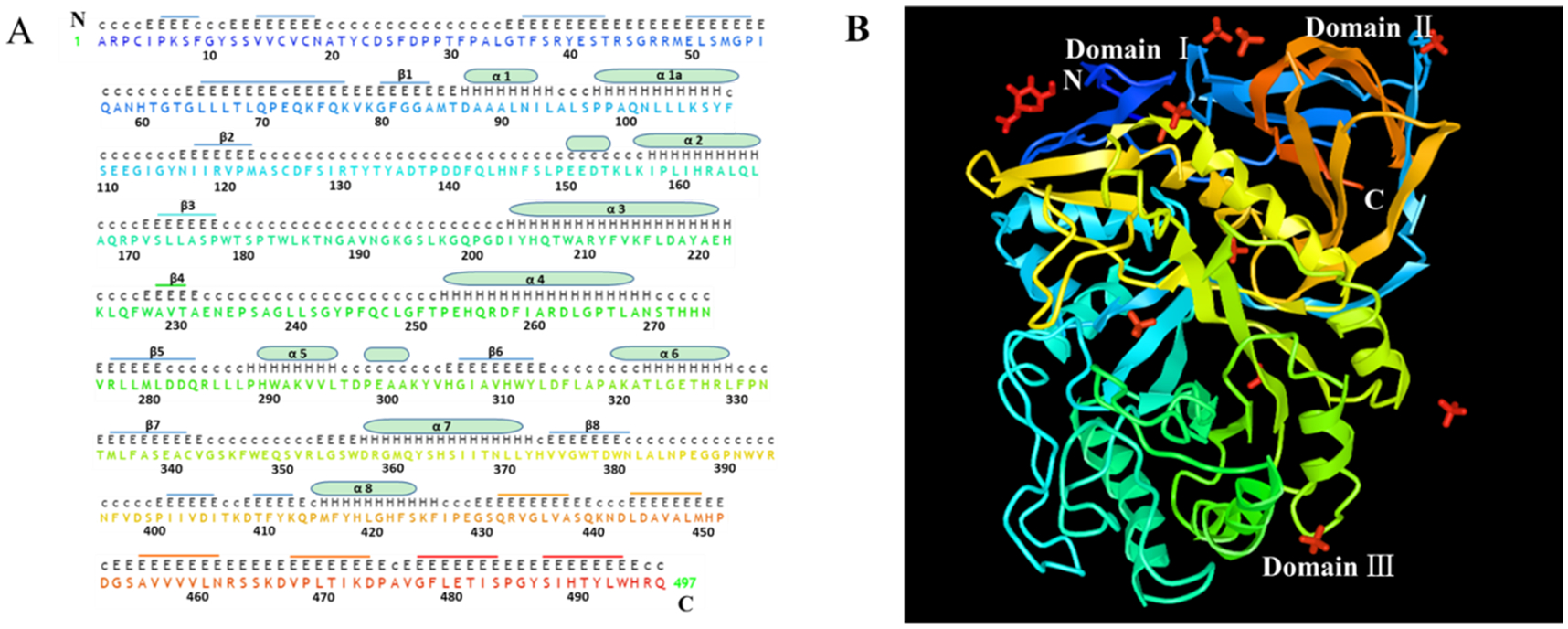
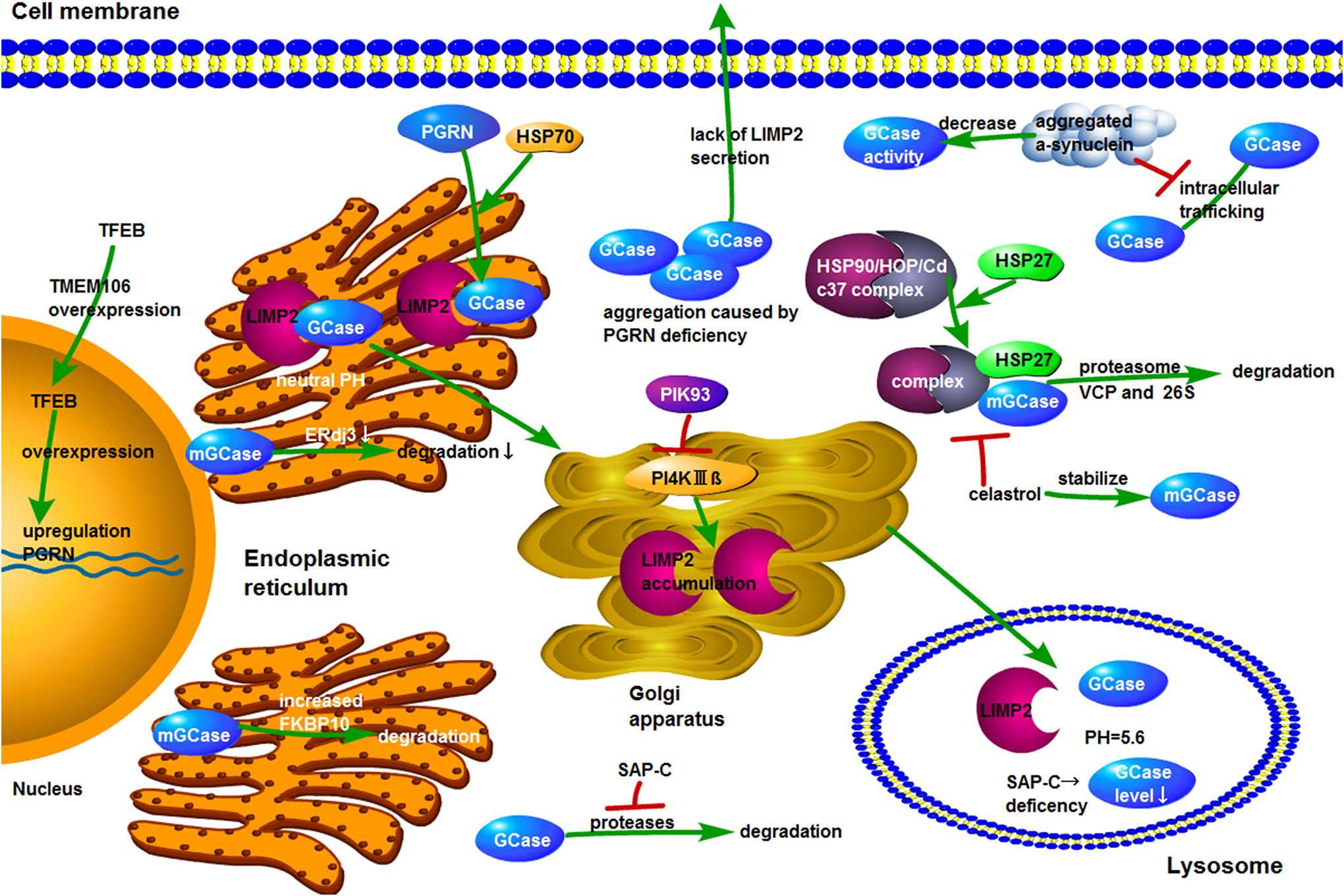
References
-
- Barkhuizen M, Anderson DG and Grobler AF, Advances in GBA-associated Parkinson’s disease--Pathology, presentation and therapies, Neurochem Int. 93 (2016) 6–25. - PubMed
-
- Beutler E, Gelbart T and Scott CR, Hematologically important mutations: Gaucher disease, Blood Cells Mol Dis. 35 (2005) 355–64. - PubMed
-
- Grabowski GA and Horowitz M, Gaucher’s disease: molecular, genetic and enzymological aspects, Baillieres Clin Haematol. 10 (1997) 635–56. - PubMed
-
- Adar T, Ilan Y, Elstein D and Zimran A, Liver involvement in Gaucher disease - Review and clinical approach, Blood Cells Mol Dis. 68 (2018) 66–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
