

TRAIL mediates and sustains constitutive NF-κB activation in LGL leukemia
- PMID: 29699990
- PMCID: PMC6014359
- DOI: 10.1182/blood-2017-09-808816
TRAIL mediates and sustains constitutive NF-κB activation in LGL leukemia
Abstract
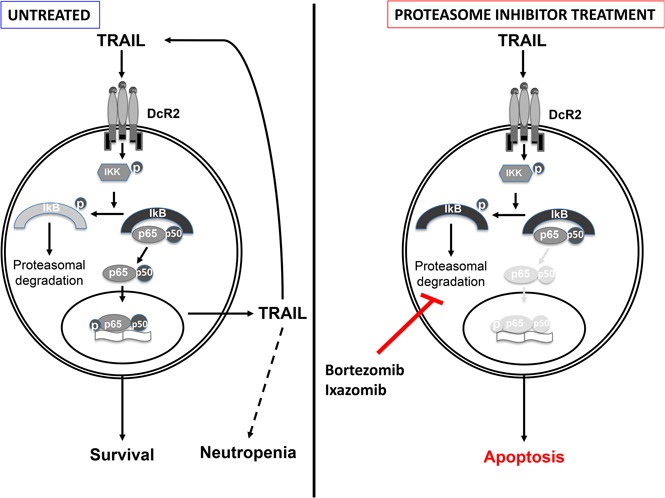
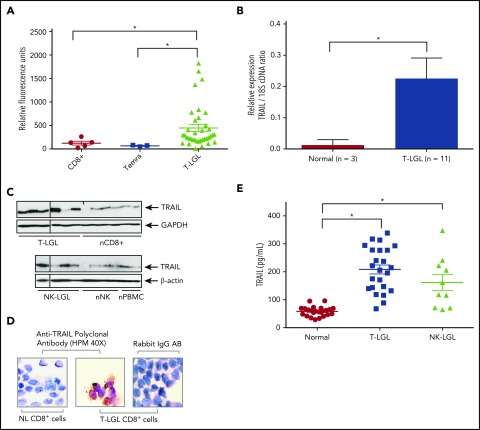
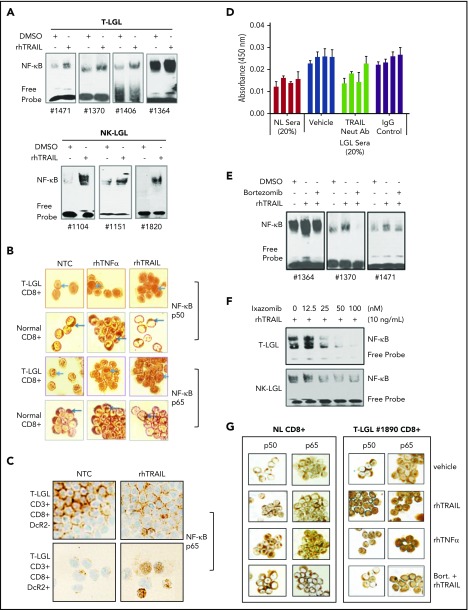
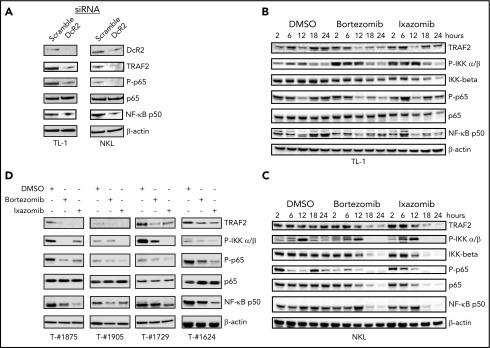
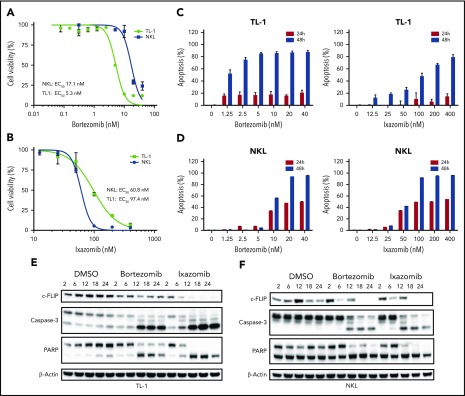
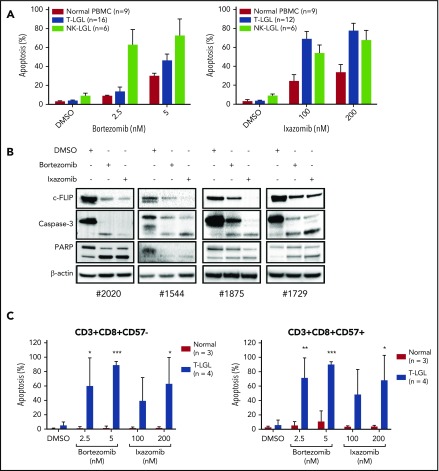
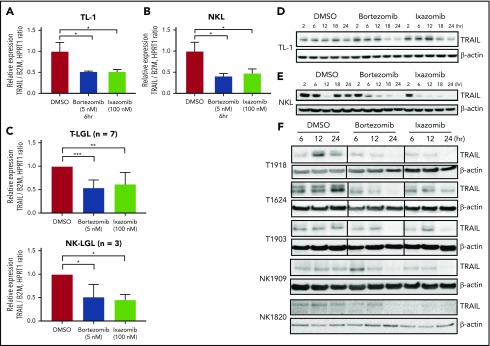
Large granular lymphocyte (LGL) leukemia results from clonal expansion of CD3+ cytotoxic T lymphocytes or CD3- natural killer (NK) cells. Chronic antigen stimulation is postulated to promote long-term survival of LGL leukemia cells through constitutive activation of multiple survival pathways, resulting in global dysregulation of apoptosis and resistance to activation-induced cell death. We reported previously that nuclear factor κB (NF-κB) is a central regulator of the survival network for leukemic LGL. However, the mechanisms that trigger constitutive activation of NF-κB in LGL leukemia remain undefined. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is known to induce apoptosis in tumor cells but can also activate NF-κB through interaction with TRAIL receptors 1, 2, and 4 (also known as DR4, DR5, and DcR2, respectively). The role of TRAIL has not been studied in LGL leukemia. In this study, we hypothesized that TRAIL interaction with DcR2 contributes to NF-κB activation in LGL leukemia. We observed upregulated TRAIL messenger RNA and protein expression in LGL leukemia cells with elevated levels of soluble TRAIL protein in LGL leukemia patient sera. We also found that DcR2 is the predominant TRAIL receptor in LGL leukemia cells. We demonstrated that TRAIL-induced activation of DcR2 led to increased NF-κB activation in leukemic LGL. Conversely, interruption of TRAIL-DcR2 signaling led to decreased NF-κB activation. Finally, a potential therapeutic application of proteasome inhibitors (bortezomib and ixazomib), which are known to inhibit NF-κB, was identified through their ability to decrease proliferation and increase apoptosis in LGL leukemia cell lines and primary patient cells.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures

References
-
- Loughran TP., Jr Clonal diseases of large granular lymphocytes. Blood. 1993;82(1):1-14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
