

Autophagy regulates exosomal release of prions in neuronal cells
- PMID: 29700113
- PMCID: PMC5995502
- DOI: 10.1074/jbc.RA117.000713
Autophagy regulates exosomal release of prions in neuronal cells
Abstract
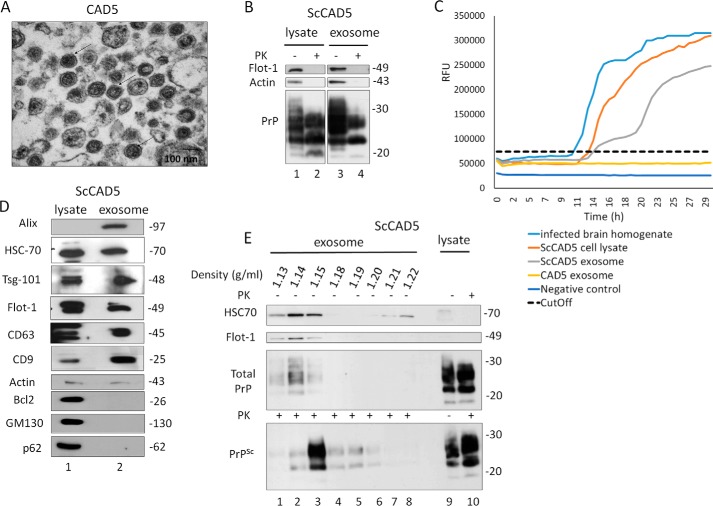
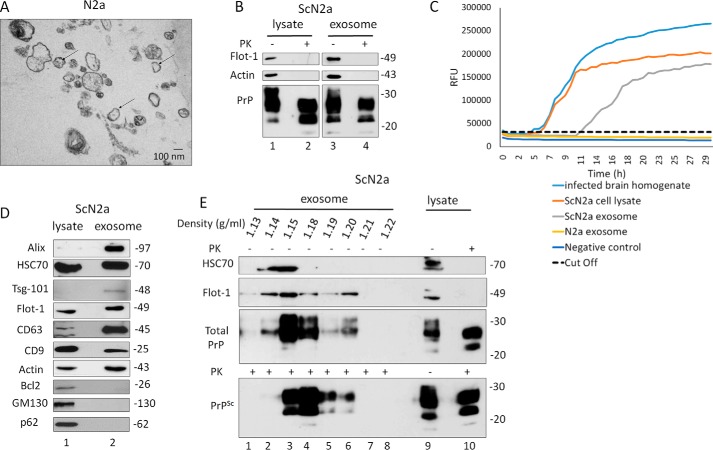
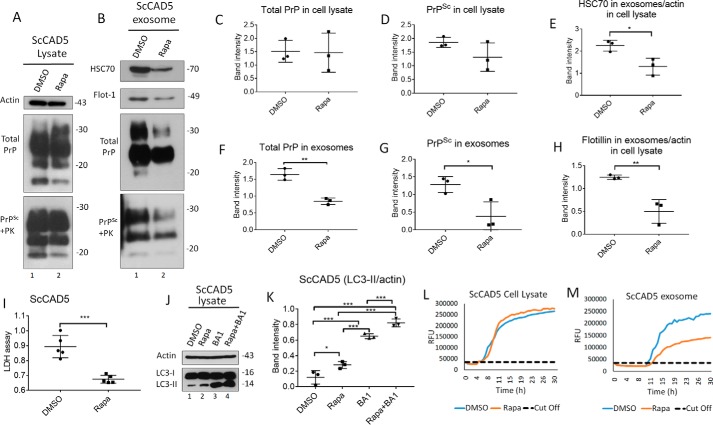
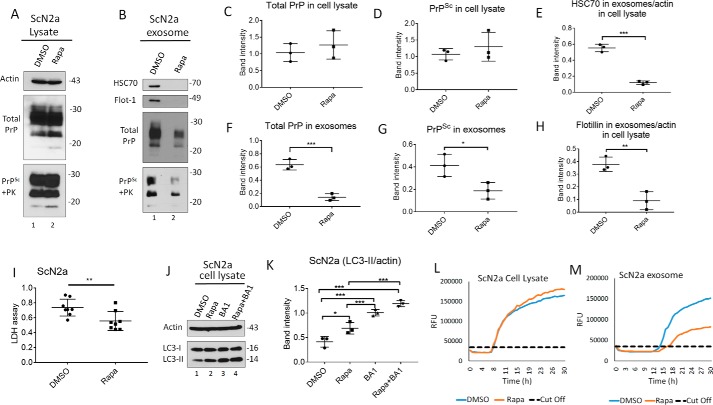
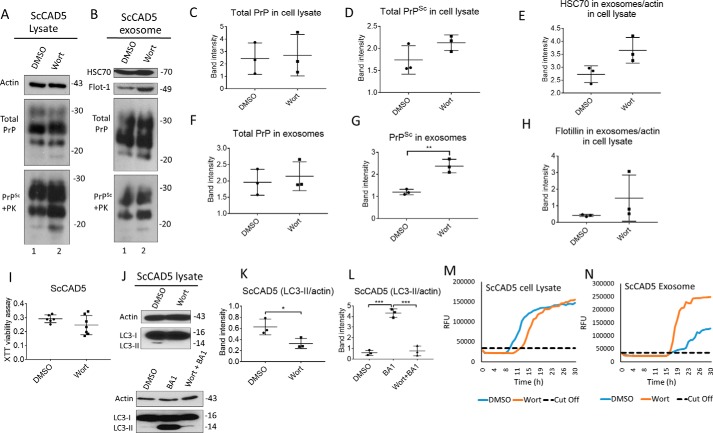
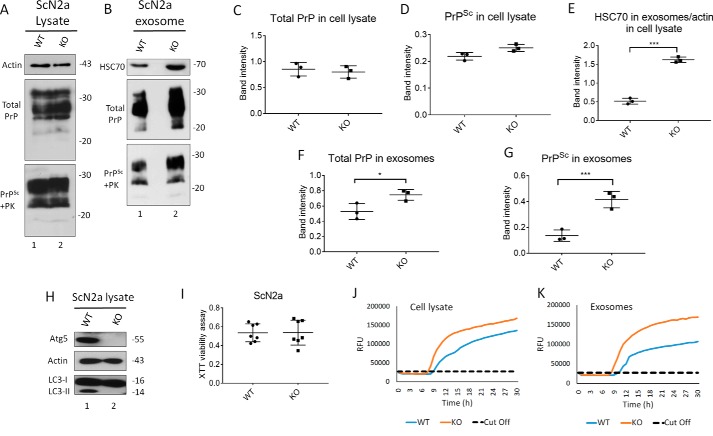
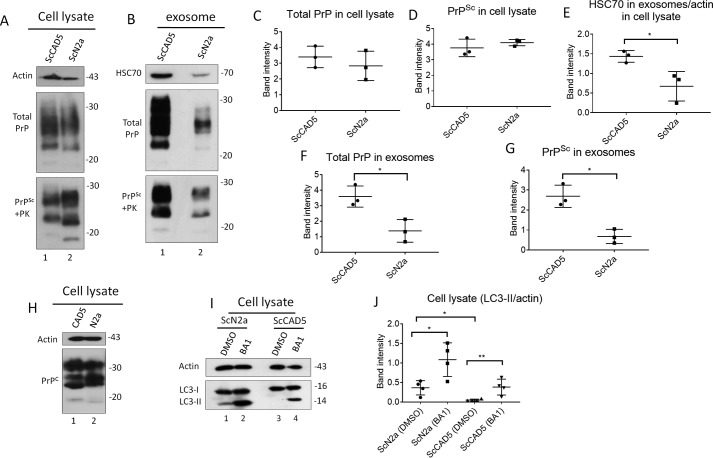
Prions are protein-based infectious agents that autocatalytically convert the cellular prion protein PrPC to its pathological isoform PrPSc Subsequent aggregation and accumulation of PrPSc in nervous tissues causes several invariably fatal neurodegenerative diseases in humans and animals. Prions can infect recipient cells when packaged into endosome-derived nanoparticles called exosomes, which are present in biological fluids such as blood, urine, and saliva. Autophagy is a basic cellular degradation and recycling machinery that also affects exosomal processing, but whether autophagy controls release of prions in exosomes is unclear. Our work investigated the effect of autophagy modulation on exosomal release of prions and how this interplay affects cellular prion infection. Exosomes isolated from cultured murine central neuronal cells (CAD5) and peripheral neuronal cells (N2a) contained prions as shown by immunoblotting for PrPSc, prion-conversion activity, and cell culture infection. We observed that autophagy stimulation with the mTOR inhibitor rapamycin strongly inhibited exosomal prion release. In contrast, inhibition of autophagy by wortmannin or CRISPR/Cas9-mediated knockout of the autophagy protein Atg5 (autophagy-related 5) greatly increased the release of exosomes and exosome-associated prions. We also show that a difference in exosomal prion release between CAD5 and N2a cells is related to differences at the level of basal autophagy. Taken together, our results indicate that autophagy modulation can control lateral transfer of prions by interfering with their exosomal release. We describe a novel role of autophagy in the prion life cycle, an understanding that may provide useful targets for containing prion diseases.
Keywords: CRISPR/Cas; Creutzfeldt–Jakob disease; RT-QuIC; autophagy; exosome (vesicle); extracellular vesicles; neurodegeneration; prion; prion disease; scrapie.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
