

Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis
- PMID: 29700225
- PMCID: PMC6247916
- DOI: 10.1126/science.aar3131
Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis
Abstract
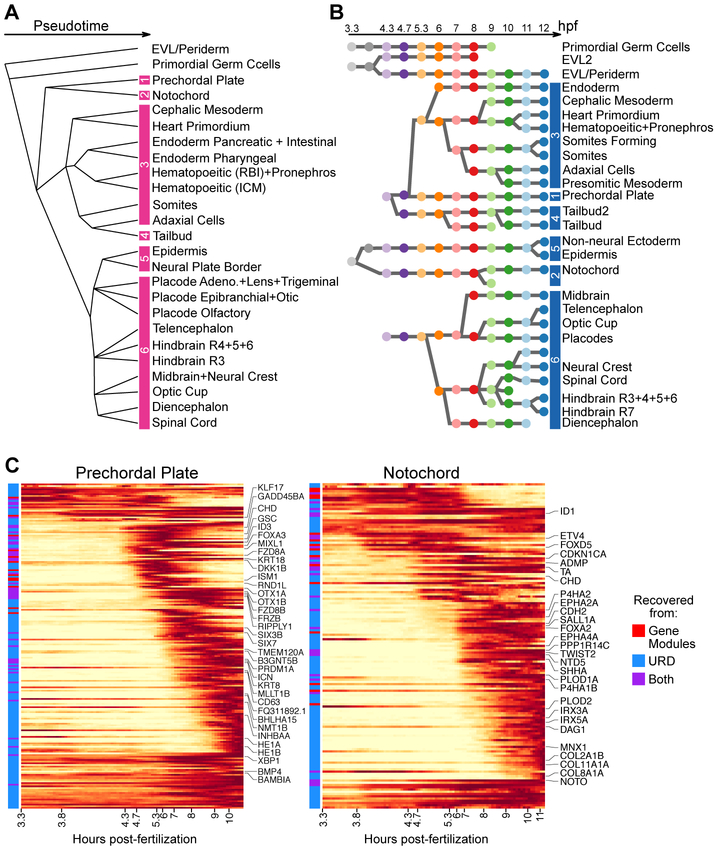
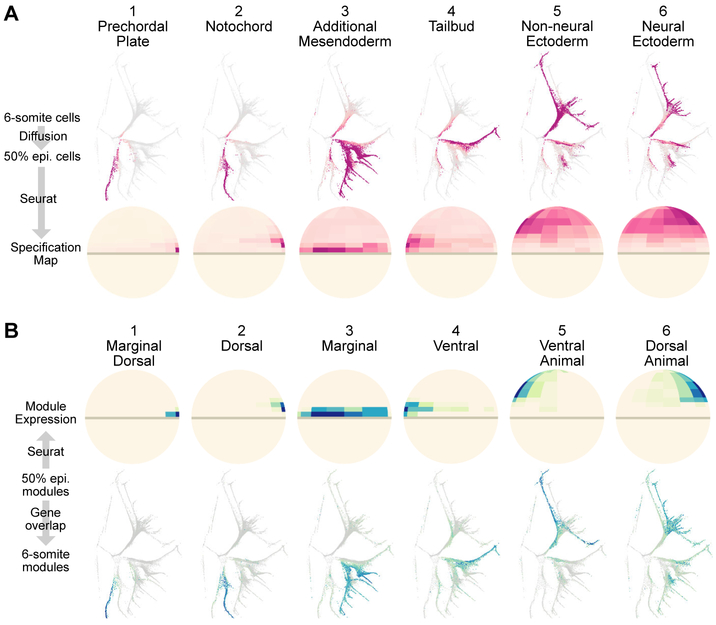
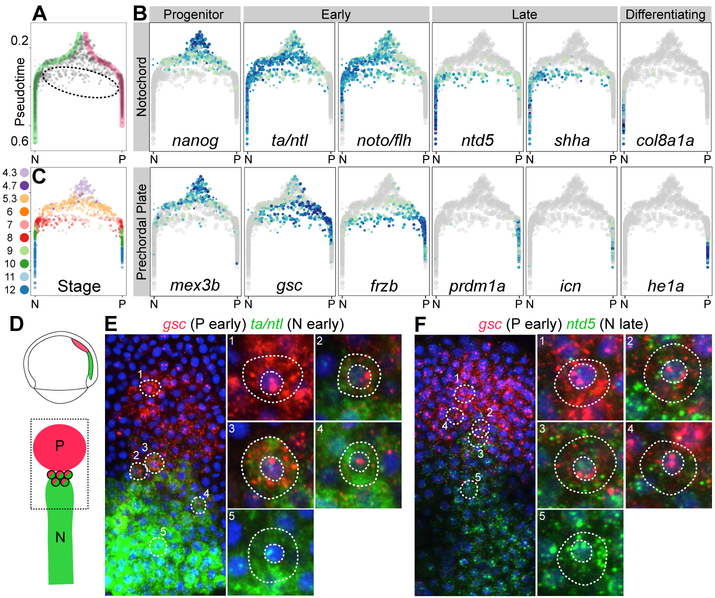
During embryogenesis, cells acquire distinct fates by transitioning through transcriptional states. To uncover these transcriptional trajectories during zebrafish embryogenesis, we sequenced 38,731 cells and developed URD, a simulated diffusion-based computational reconstruction method. URD identified the trajectories of 25 cell types through early somitogenesis, gene expression along them, and their spatial origin in the blastula. Analysis of Nodal signaling mutants revealed that their transcriptomes were canalized into a subset of wild-type transcriptional trajectories. Some wild-type developmental branch points contained cells that express genes characteristic of multiple fates. These cells appeared to trans-specify from one fate to another. These findings reconstruct the transcriptional trajectories of a vertebrate embryo, highlight the concurrent canalization and plasticity of embryonic specification, and provide a framework with which to reconstruct complex developmental trees from single-cell transcriptomes.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures



Comment in
-
A new view of embryo development and regeneration.Science. 2018 Jun 1;360(6392):967-968. doi: 10.1126/science.aat8413. Science. 2018. PMID: 29853675 No abstract available.
References
-
- Davis RL, Weintraub H, Lassar AB, Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 51, 987–1000 (1987). - PubMed
-
- Tapscott SJ et al., MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 242, 405–411 (1988). - PubMed
-
- Takahashi K, Yamanaka S, Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 126, 663–676 (2006). - PubMed
-
- Cannoodt R, Saelens W, Saeys Y, Computational methods for trajectory inference from single-cell transcriptomics. Eur. J. Immunol 46, 2496–2506 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
