

KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery
- PMID: 29700325
- PMCID: PMC5920042
- DOI: 10.1038/s41598-018-24900-3
KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery
Abstract
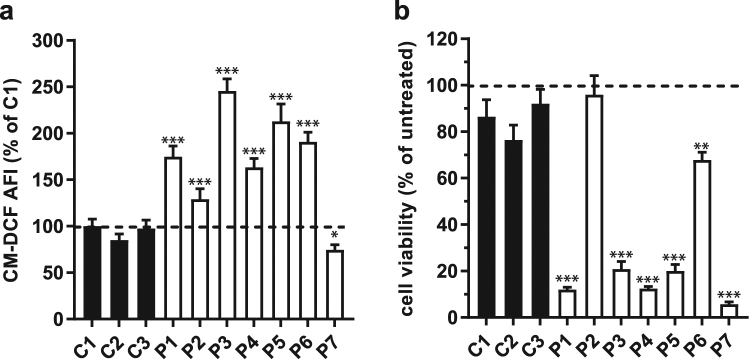
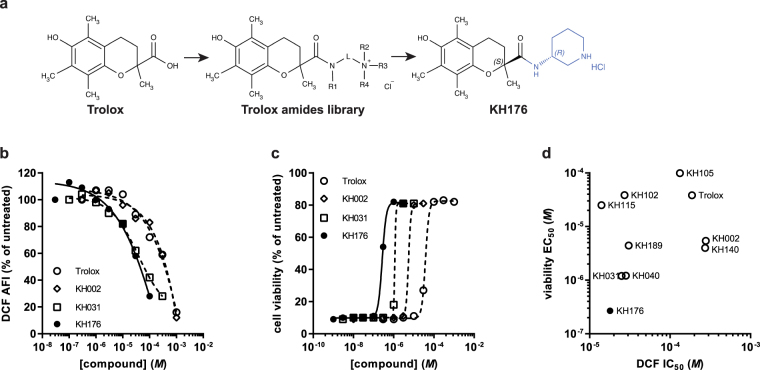
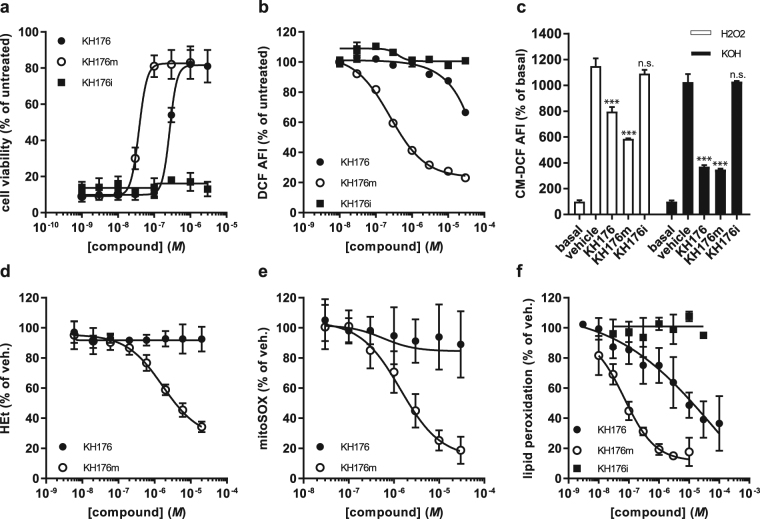
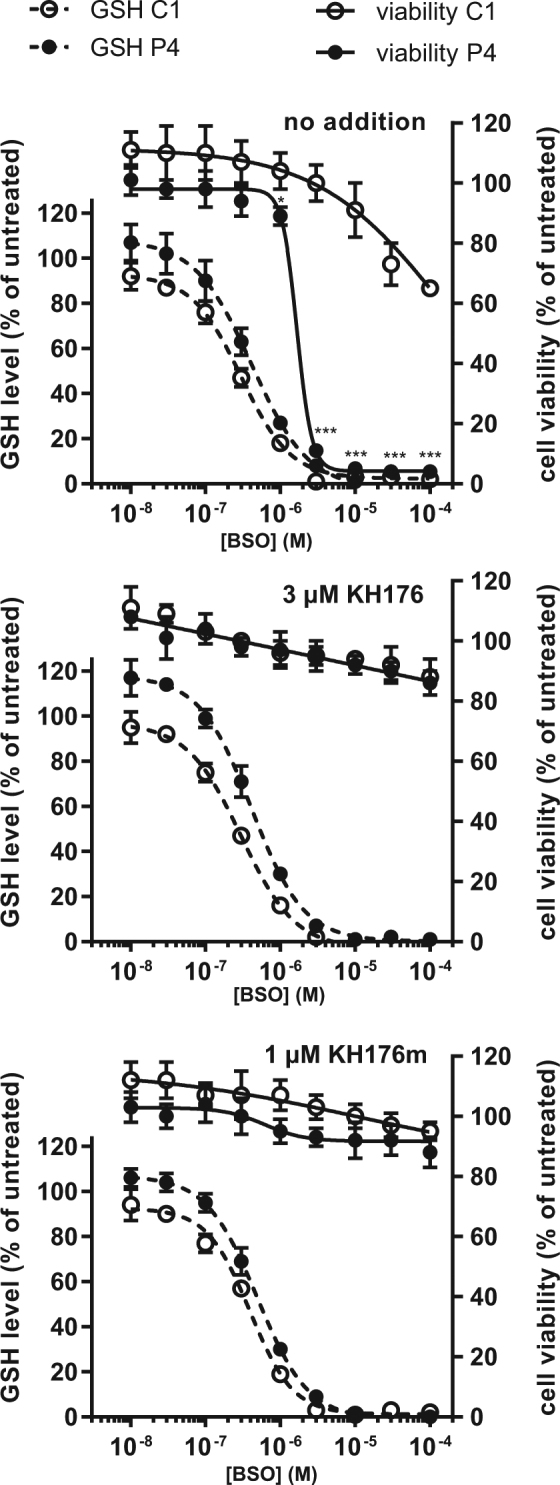
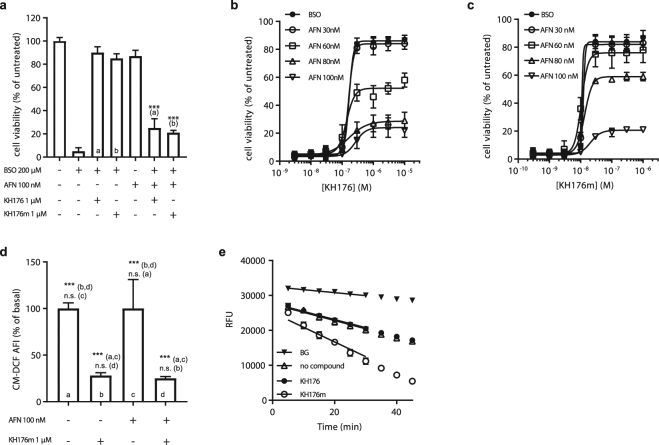
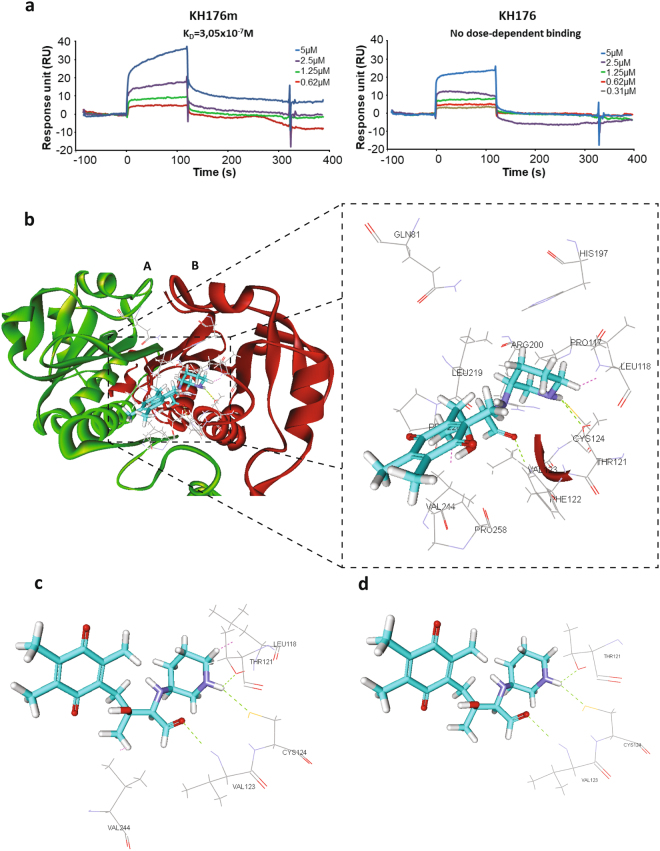
A deficient activity of one or more of the mitochondrial oxidative phosphorylation (OXPHOS) enzyme complexes leads to devastating diseases, with high unmet medical needs. Mitochondria, and more specifically the OXPHOS system, are the main cellular production sites of Reactive Oxygen Species (ROS). Increased ROS production, ultimately leading to irreversible oxidative damage of macromolecules or to more selective and reversible redox modulation of cell signalling, is a causative hallmark of mitochondrial diseases. Here we report on the development of a new clinical-stage drug KH176 acting as a ROS-Redox modulator. Patient-derived primary skin fibroblasts were used to assess the potency of a new library of chromanyl-based compounds to reduce ROS levels and protect cells against redox-stress. The lead compound KH176 was studied in cell-based and enzymatic assays and in silico. Additionally, the metabolism, pharmacokinetics and toxicokinetics of KH176 were assessed in vivo in different animal species. We demonstrate that KH176 can effectively reduce increased cellular ROS levels and protect OXPHOS deficient primary cells against redox perturbation by targeting the Thioredoxin/Peroxiredoxin system. Due to its dual activity as antioxidant and redox modulator, KH176 offers a novel approach to the treatment of mitochondrial (-related) diseases. KH176 efficacy and safety are currently being evaluated in a Phase 2 clinical trial.
Conflict of interest statement
J.B., H.R., M.P., L.H., S.P., P.Z. are fully employed by Khondrion. J.S. is the founding CEO of Khondrion. A.B. is fully employed by Inoviem Scientific. P.E. is the founding CEO of Inoviem Scientific.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
