

An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder
- PMID: 29700473
- PMCID: PMC5961723
- DOI: 10.1038/s41588-018-0107-y
An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder
Abstract
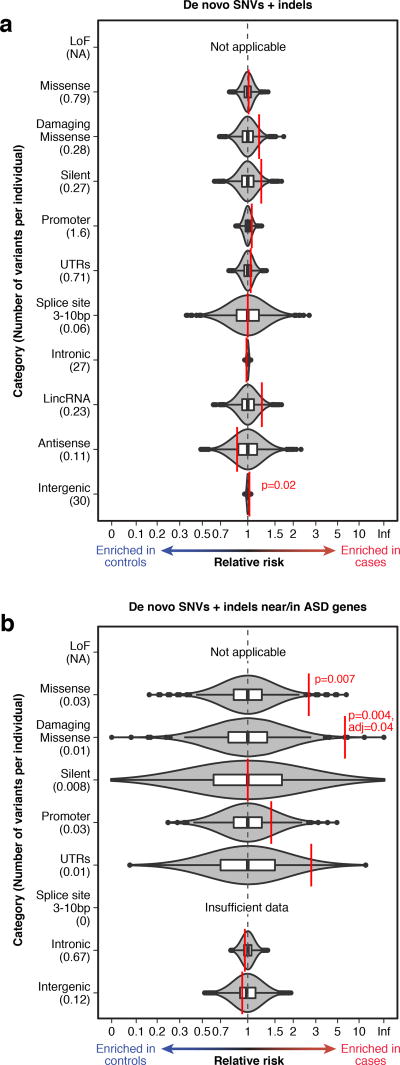
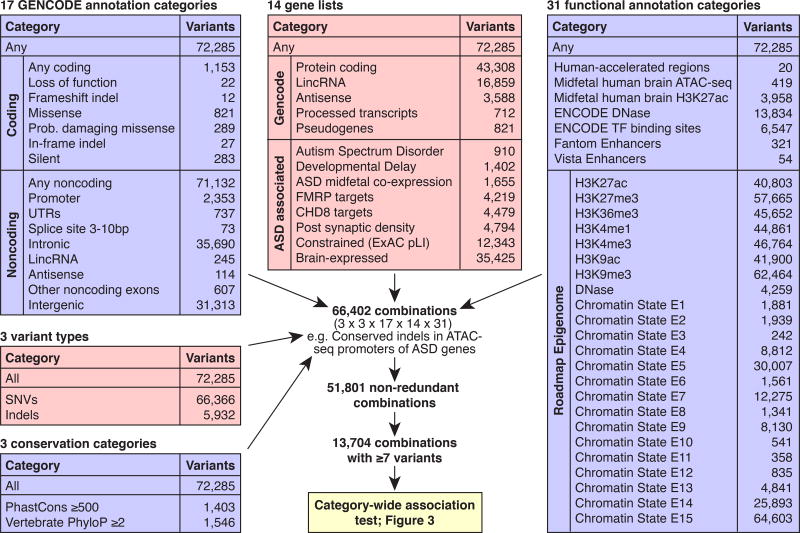
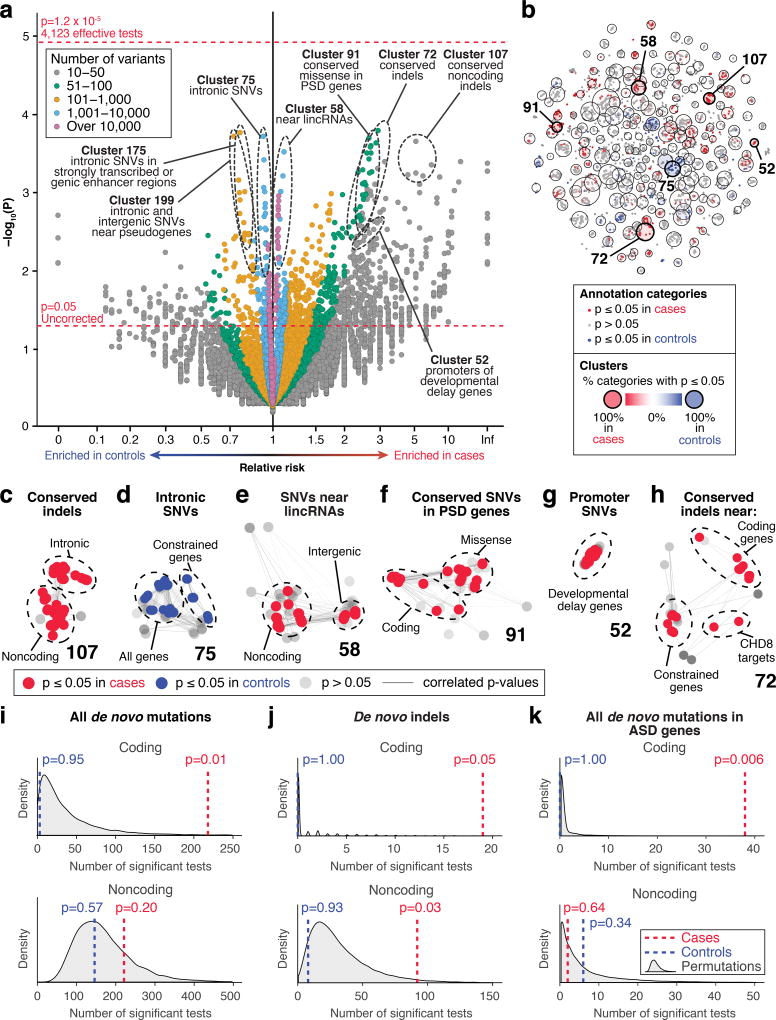
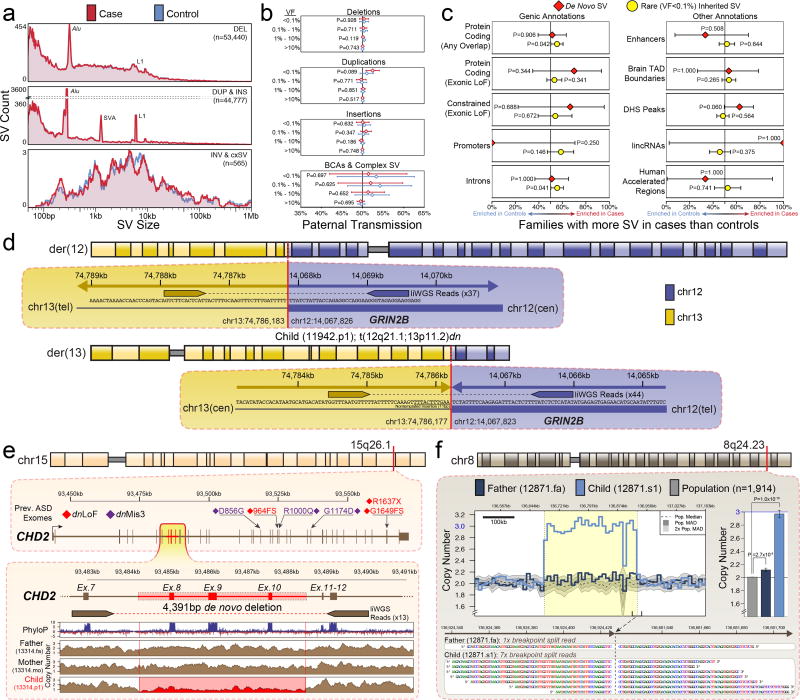
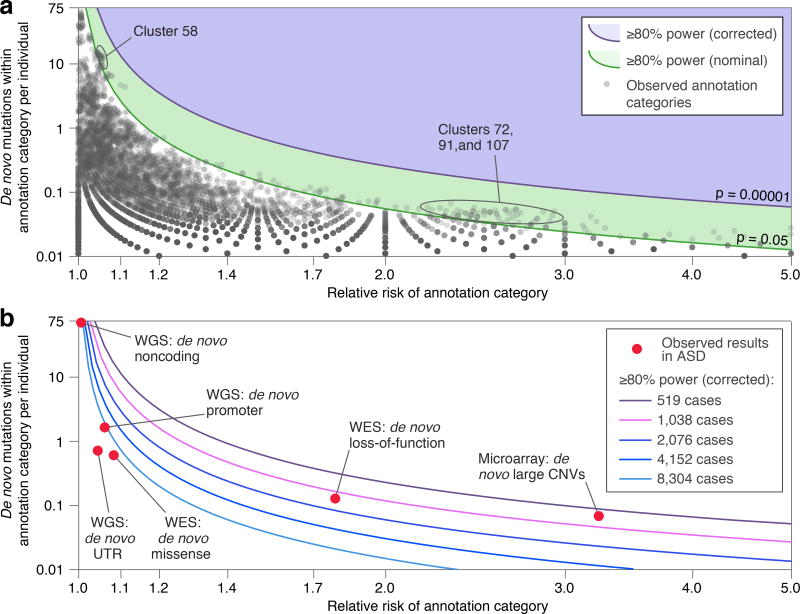
Genomic association studies of common or rare protein-coding variation have established robust statistical approaches to account for multiple testing. Here we present a comparable framework to evaluate rare and de novo noncoding single-nucleotide variants, insertion/deletions, and all classes of structural variation from whole-genome sequencing (WGS). Integrating genomic annotations at the level of nucleotides, genes, and regulatory regions, we define 51,801 annotation categories. Analyses of 519 autism spectrum disorder families did not identify association with any categories after correction for 4,123 effective tests. Without appropriate correction, biologically plausible associations are observed in both cases and controls. Despite excluding previously identified gene-disrupting mutations, coding regions still exhibited the strongest associations. Thus, in autism, the contribution of de novo noncoding variation is probably modest in comparison to that of de novo coding variants. Robust results from future WGS studies will require large cohorts and comprehensive analytical strategies that consider the substantial multiple-testing burden.
Figures
Comment in
-
Straws in a haystack.Nat Genet. 2018 May;50(5):631. doi: 10.1038/s41588-018-0125-9. Nat Genet. 2018. PMID: 29700466 No abstract available.
-
Sizing up whole-genome sequencing studies of common diseases.Nat Genet. 2018 May;50(5):635-637. doi: 10.1038/s41588-018-0113-0. Nat Genet. 2018. PMID: 29700468 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 MH110928/MH/NIMH NIH HHS/United States
- R37 MH057881/MH/NIMH NIH HHS/United States
- U01 MH105575/MH/NIMH NIH HHS/United States
- U01 MH100229/MH/NIMH NIH HHS/United States
- K99 DE026824/DE/NIDCR NIH HHS/United States
- U01 MH111660/MH/NIMH NIH HHS/United States
- U01 MH100239/MH/NIMH NIH HHS/United States
- R35 NS097305/NS/NINDS NIH HHS/United States
- U01 MH111658/MH/NIMH NIH HHS/United States
- U01 MH111661/MH/NIMH NIH HHS/United States
- P01 GM061354/GM/NIGMS NIH HHS/United States
- R01 MH109901/MH/NIMH NIH HHS/United States
- R01 HD081256/HD/NICHD NIH HHS/United States
- U01 MH111662/MH/NIMH NIH HHS/United States
- R01 MH109904/MH/NIMH NIH HHS/United States
- R01 MH109900/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
