

Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits
- PMID: 29700474
- PMCID: PMC5934350
- DOI: 10.1038/s41588-018-0108-x
Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits
Abstract
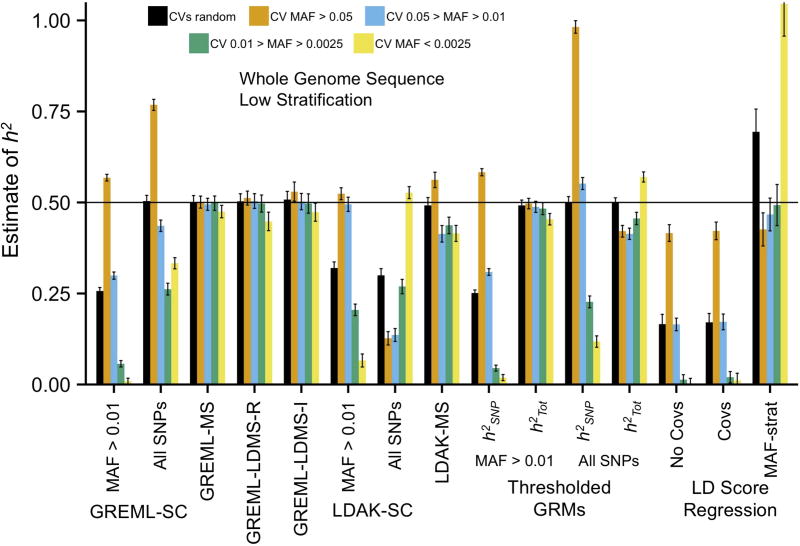
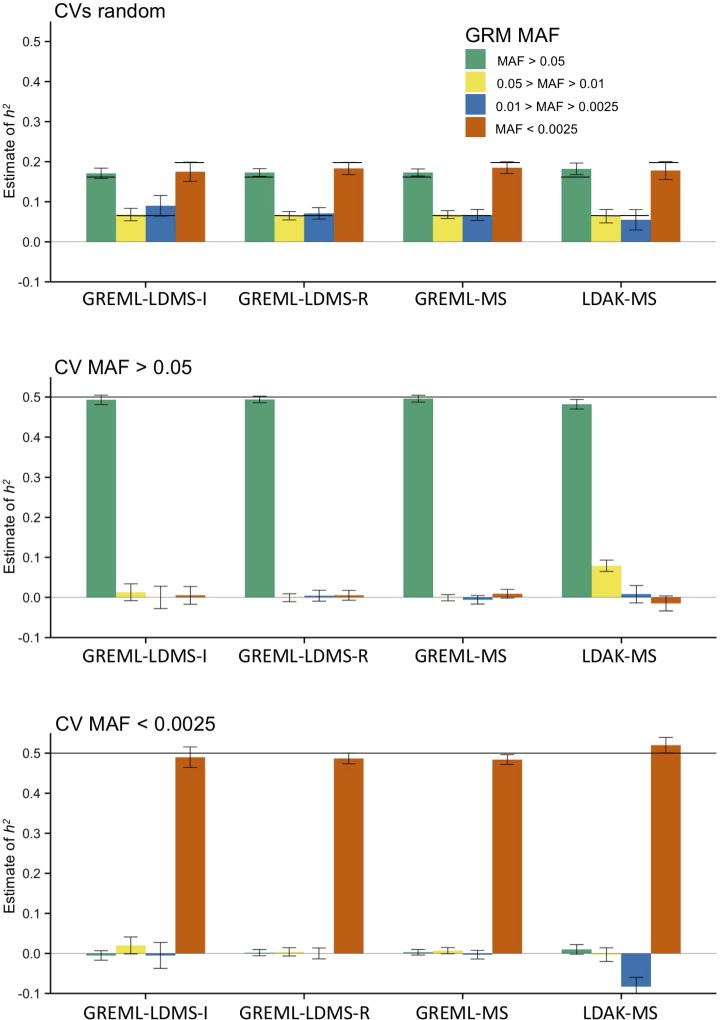
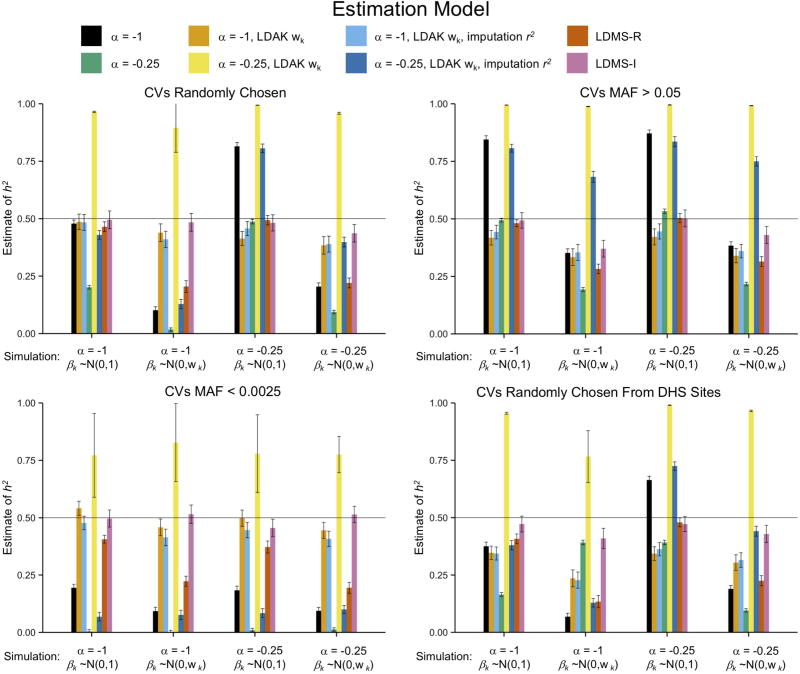
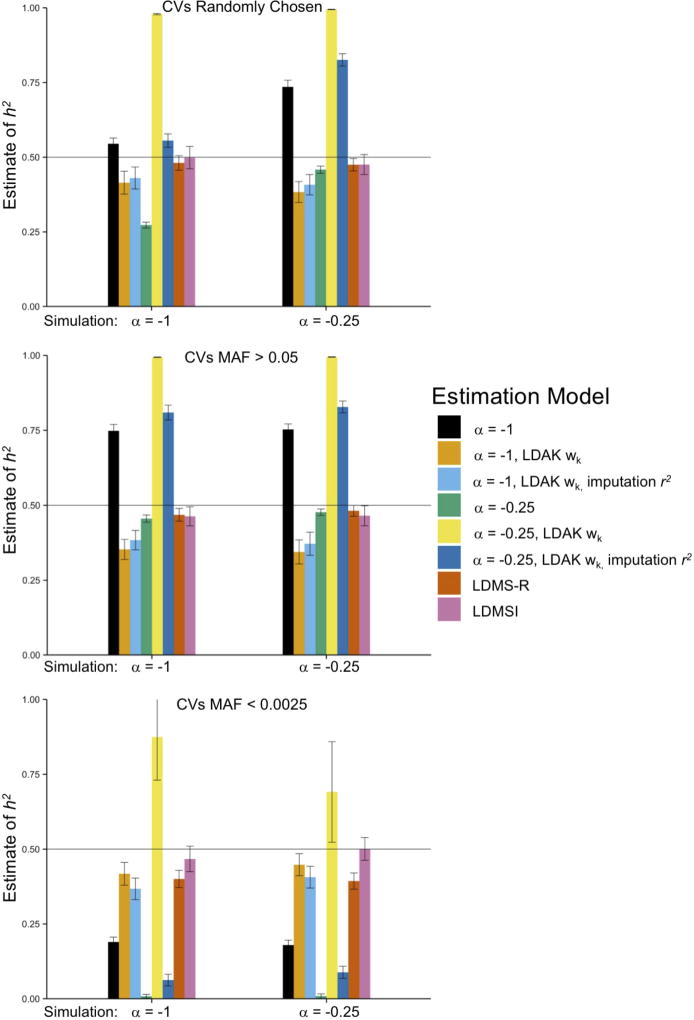
Multiple methods have been developed to estimate narrow-sense heritability, h2, using single nucleotide polymorphisms (SNPs) in unrelated individuals. However, a comprehensive evaluation of these methods has not yet been performed, leading to confusion and discrepancy in the literature. We present the most thorough and realistic comparison of these methods to date. We used thousands of real whole-genome sequences to simulate phenotypes under varying genetic architectures and confounding variables, and we used array, imputed, or whole genome sequence SNPs to obtain 'SNP-heritability' estimates. We show that SNP-heritability can be highly sensitive to assumptions about the frequencies, effect sizes, and levels of linkage disequilibrium of underlying causal variants, but that methods that bin SNPs according to minor allele frequency and linkage disequilibrium are less sensitive to these assumptions across a wide range of genetic architectures and possible confounding factors. These findings provide guidance for best practices and proper interpretation of published estimates.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
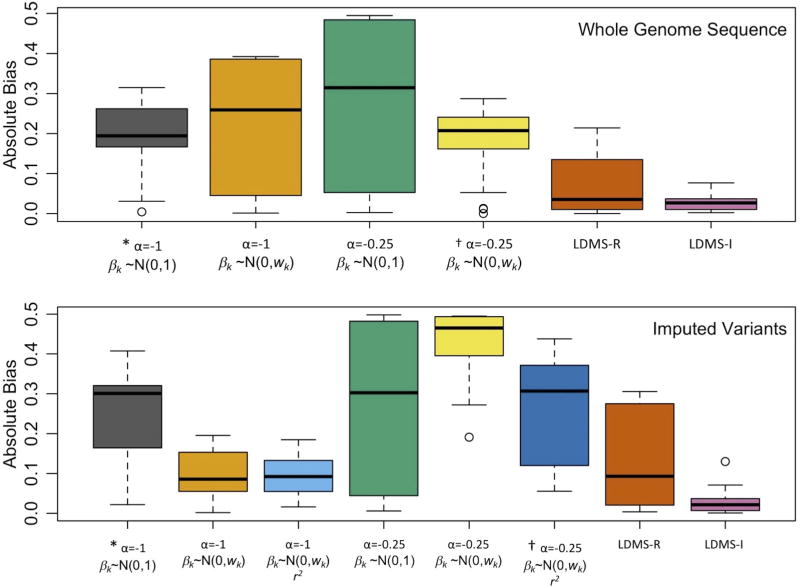
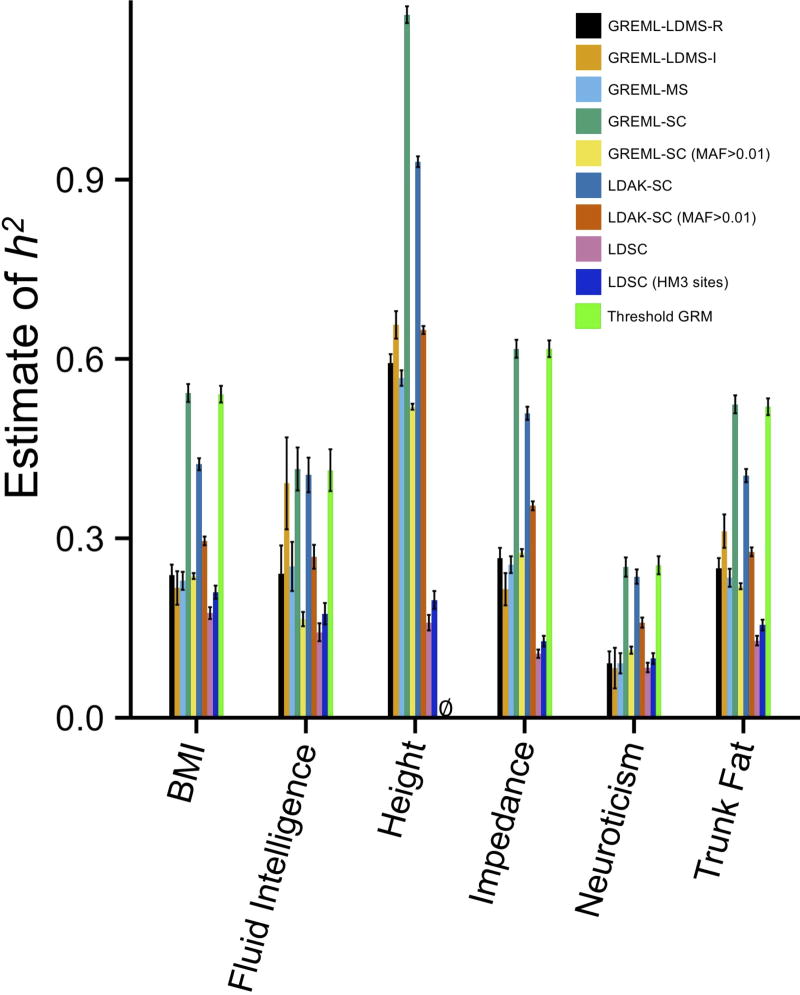
References
-
- Tenesa A, Haley CS. The heritability of human disease: estimation, uses and abuses. Nat. Rev. Genet. 2013;14:139–149. - PubMed
-
- Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat. Rev. Genet. 2008;9:255–66. - PubMed
-
- Keller MC, Coventry WL. Quantifying and addressing parameter indeterminacy in the classical twin design. Twin Res. Hum. Genet. 2005;8:201–213. - PubMed
-
- Eaves LJ, Last KA, Young PA, Martin NG. Model-fitting approaches to the analysis of human behaviour. Heredity (Edinb) 1978;41:249–320. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
