

Somatic mutations in neurons during aging and neurodegeneration
- PMID: 29705908
- PMCID: PMC5954077
- DOI: 10.1007/s00401-018-1850-y
Somatic mutations in neurons during aging and neurodegeneration
Erratum in
-
Correction to: Somatic mutations in neurons during aging and neurodegeneration.Acta Neuropathol. 2020 Sep;140(3):415. doi: 10.1007/s00401-020-02186-y. Acta Neuropathol. 2020. PMID: 32632518 Free PMC article.
Abstract
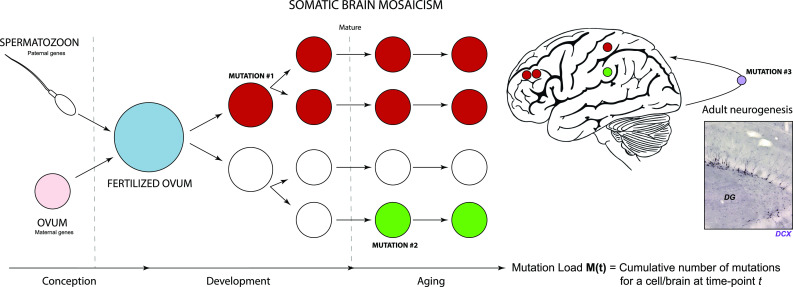
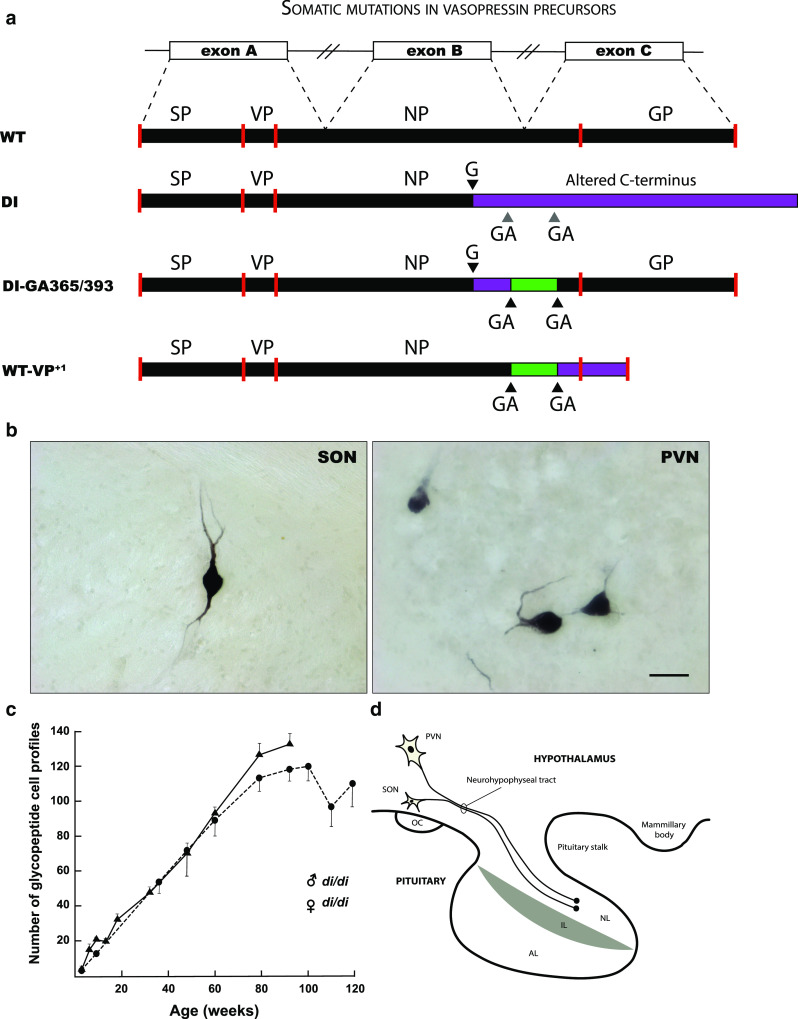
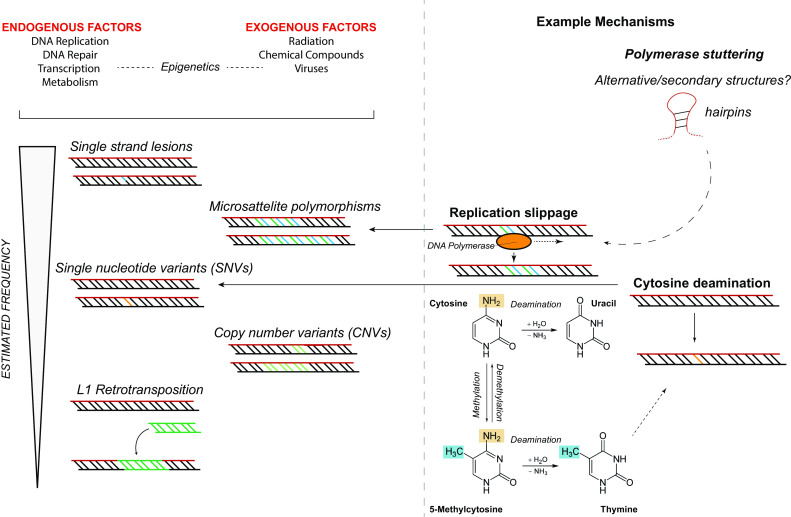
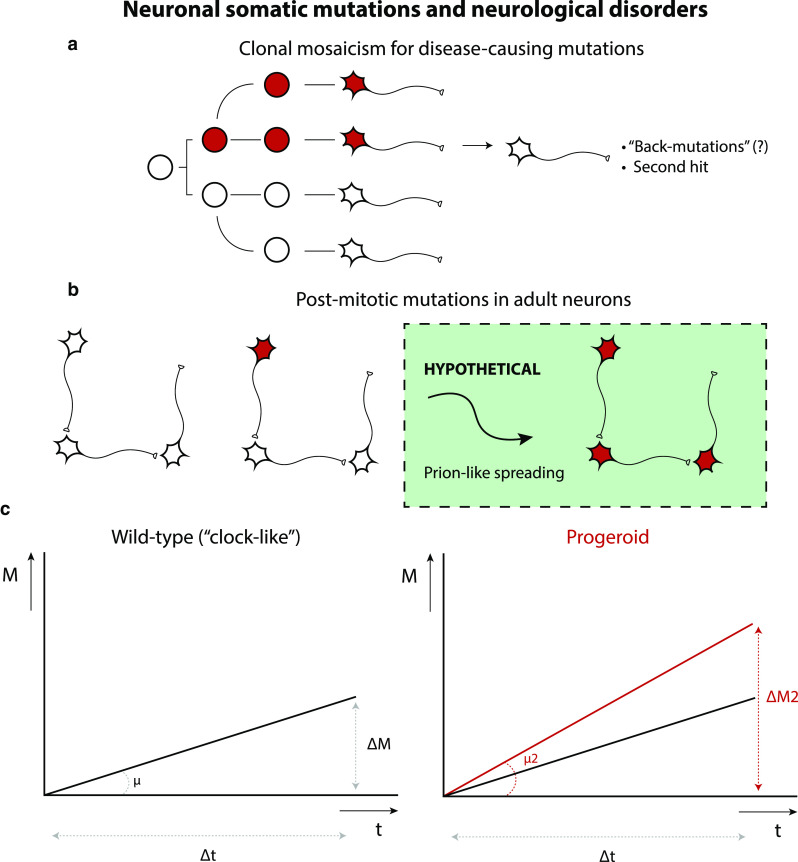
The nervous system is composed of a large variety of neurons with a diverse array of morphological and functional properties. This heterogeneity is essential for the construction and maintenance of a distinct set of neural networks with unique characteristics. Accumulating evidence now indicates that neurons do not only differ at a functional level, but also at the genomic level. These genomic discrepancies seem to be the result of somatic mutations that emerge in nervous tissue during development and aging. Ultimately, these mutations bring about a genetically heterogeneous population of neurons, a phenomenon that is commonly referred to as "somatic brain mosaicism". Improved understanding of the development and consequences of somatic brain mosaicism is crucial to understand the impact of somatic mutations on neuronal function in human aging and disease. Here, we highlight a number of topics related to somatic brain mosaicism, including some early experimental evidence for somatic mutations in post-mitotic neurons of the hypothalamo-neurohypophyseal system. We propose that age-related somatic mutations are particularly interesting, because aging is a major risk factor for a variety of neuronal diseases, including Alzheimer's disease. We highlight potential links between somatic mutations and the development of these diseases and argue that recent advances in single-cell genomics and in vivo physiology have now finally made it possible to dissect the origins and consequences of neuronal mutations in unprecedented detail.
Keywords: Aging; Genome integrity; Neurodegeneration; Neurological disorders; Neuronal development; Somatic brain mosaicism; Somatic mutations.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
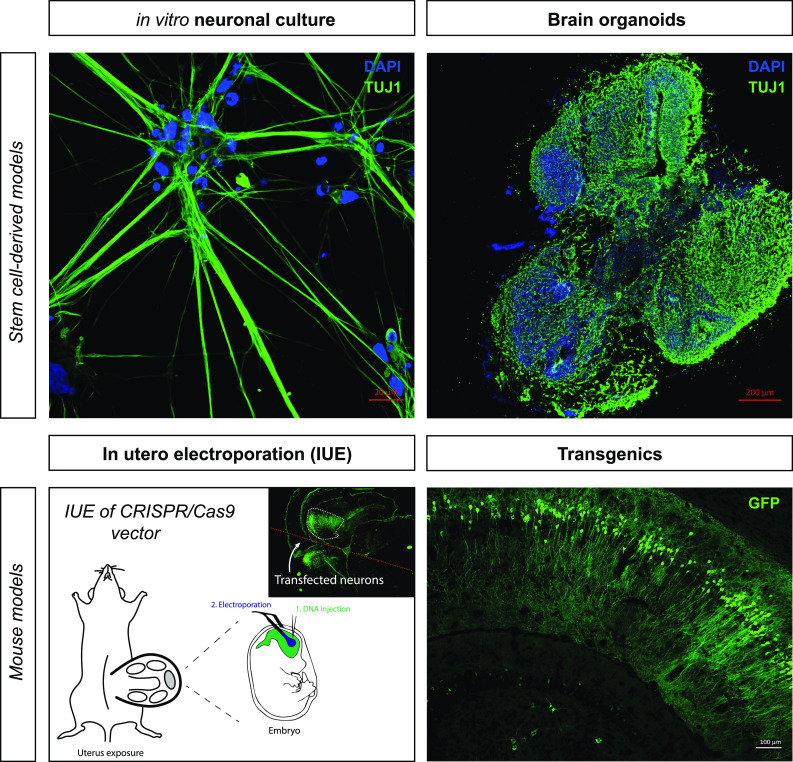
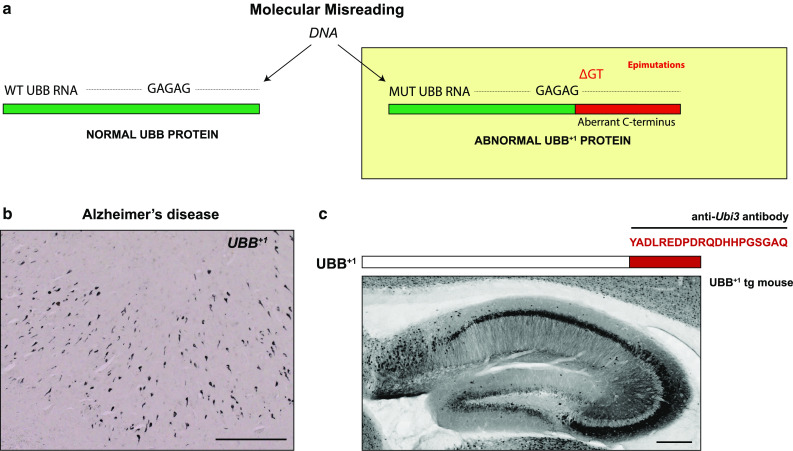
References
-
- Alzualde A, Moreno F, Martínez-Lage P, Ferrer I, Gorostidi A, Otaegui D, et al. Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo D178 N mutation in the PRNP gene. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:1283–1291. doi: 10.1002/ajmg.b.31099. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
