

Could Alzheimer's Disease Originate in the Periphery and If So How So?
- PMID: 29705945
- PMCID: PMC6372984
- DOI: 10.1007/s12035-018-1092-y
Could Alzheimer's Disease Originate in the Periphery and If So How So?
Abstract
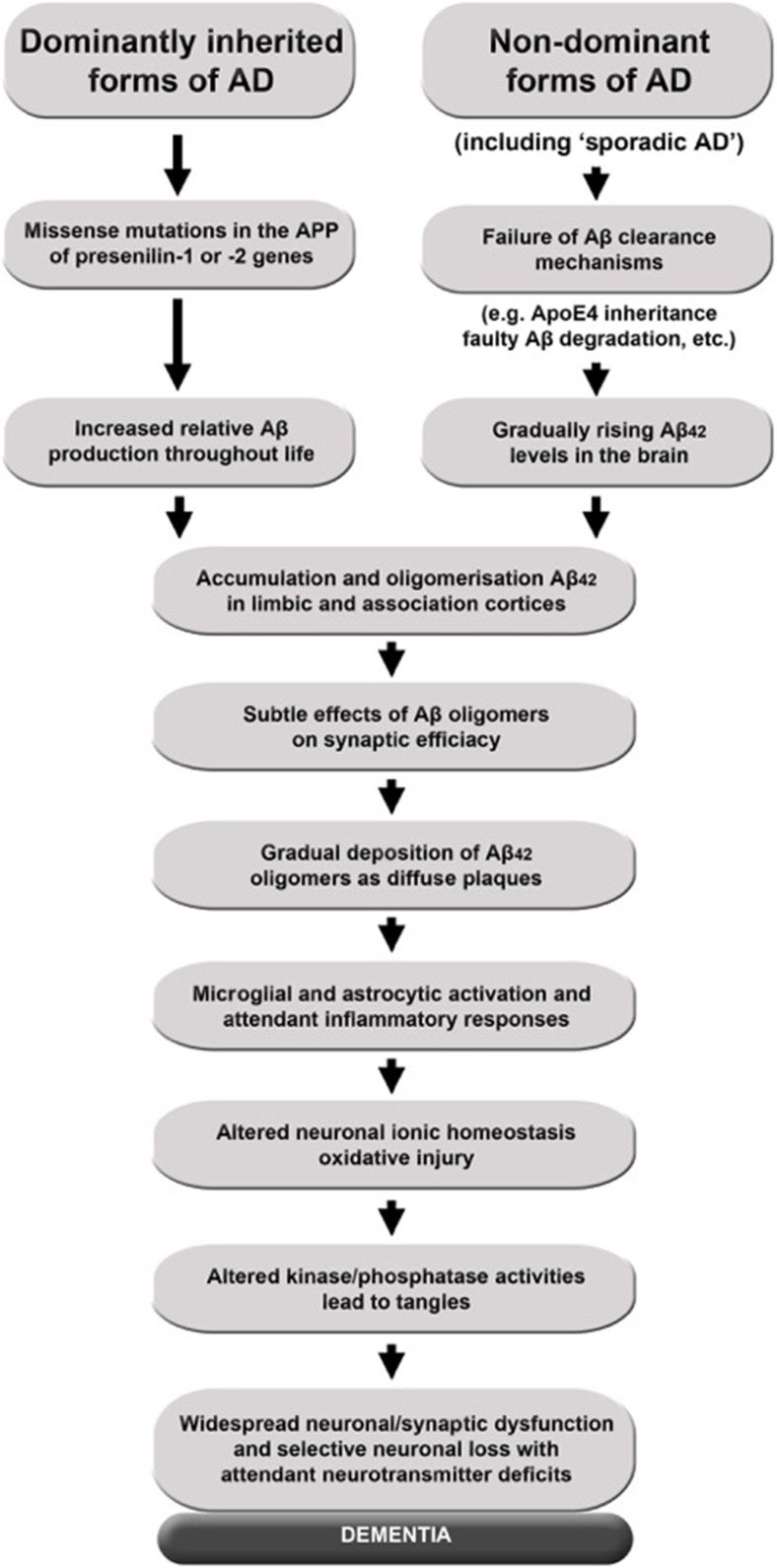
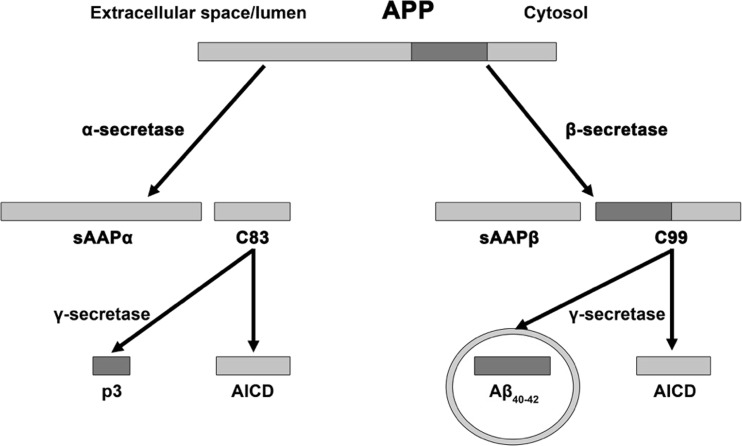
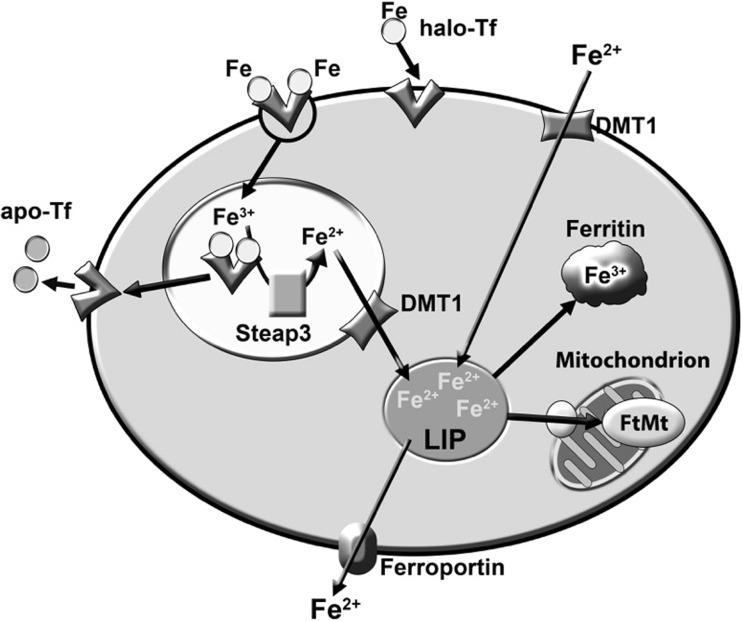
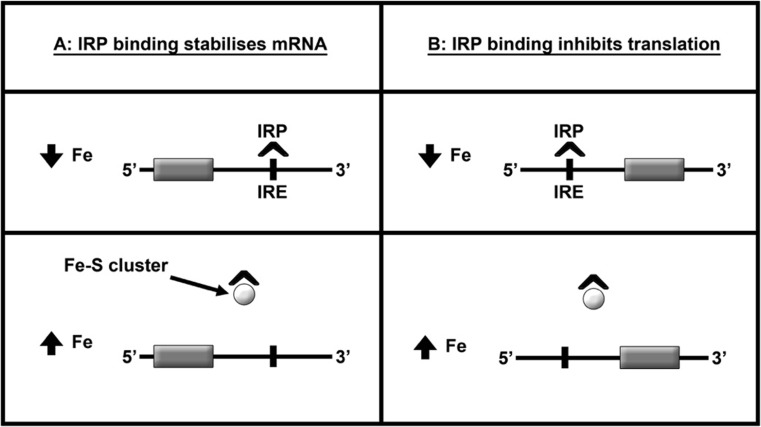
The classical amyloid cascade model for Alzheimer's disease (AD) has been challenged by several findings. Here, an alternative molecular neurobiological model is proposed. It is shown that the presence of the APOE ε4 allele, altered miRNA expression and epigenetic dysregulation in the promoter region and exon 1 of TREM2, as well as ANK1 hypermethylation and altered levels of histone post-translational methylation leading to increased transcription of TNFA, could variously explain increased levels of peripheral and central inflammation found in AD. In particular, as a result of increased activity of triggering receptor expressed on myeloid cells 2 (TREM-2), the presence of the apolipoprotein E4 (ApoE4) isoform, and changes in ANK1 expression, with subsequent changes in miR-486 leading to altered levels of protein kinase B (Akt), mechanistic (previously mammalian) target of rapamycin (mTOR) and signal transducer and activator of transcription 3 (STAT3), all of which play major roles in microglial activation, proliferation and survival, there is activation of microglia, leading to the subsequent (further) production of cytokines, chemokines, nitric oxide, prostaglandins, reactive oxygen species, inducible nitric oxide synthase and cyclooxygenase-2, and other mediators of inflammation and neurotoxicity. These changes are associated with the development of amyloid and tau pathology, mitochondrial dysfunction (including impaired activity of the electron transport chain, depleted basal mitochondrial potential and oxidative damage to key tricarboxylic acid enzymes), synaptic dysfunction, altered glycogen synthase kinase-3 (GSK-3) activity, mTOR activation, impairment of autophagy, compromised ubiquitin-proteasome system, iron dyshomeostasis, changes in APP translation, amyloid plaque formation, tau hyperphosphorylation and neurofibrillary tangle formation.
Keywords: Alzheimer’s disease; Gene expression; Inflammation; Microglia; Mitochondria; Molecular neurobiology.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
