

Pervasive Protein Thermal Stability Variation during the Cell Cycle
- PMID: 29706546
- PMCID: PMC5998384
- DOI: 10.1016/j.cell.2018.03.053
Pervasive Protein Thermal Stability Variation during the Cell Cycle
Abstract
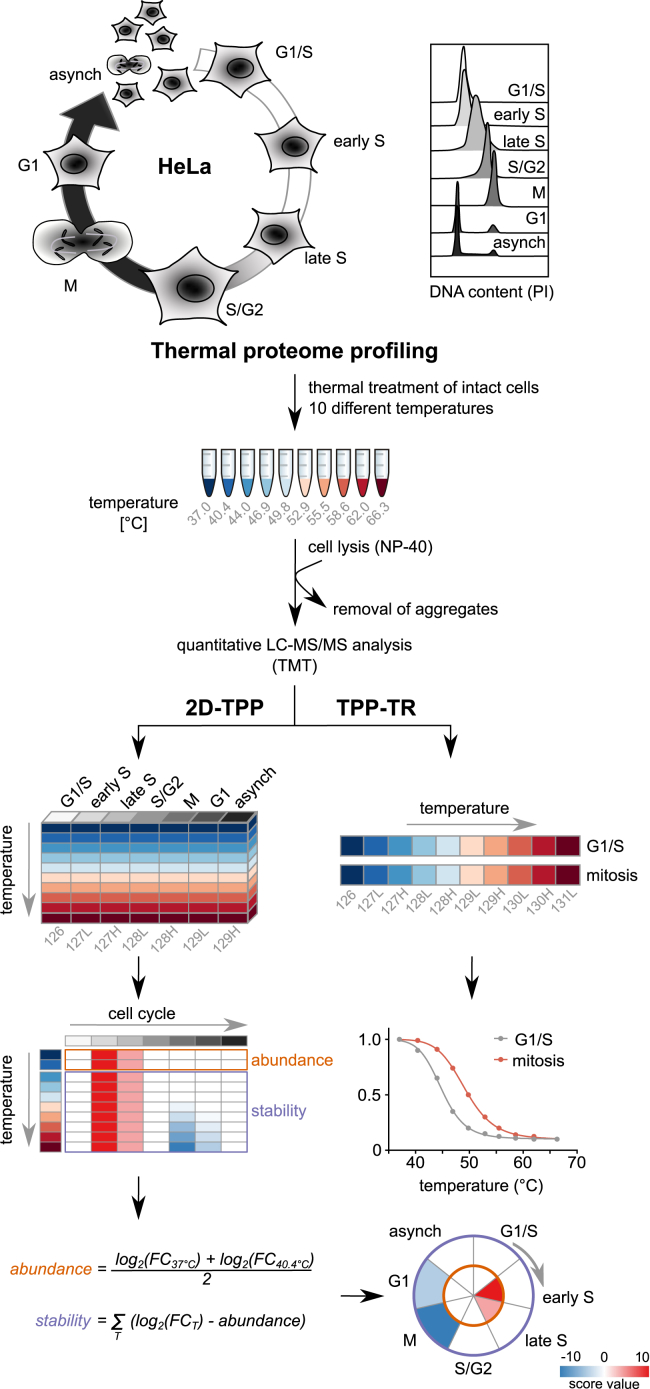
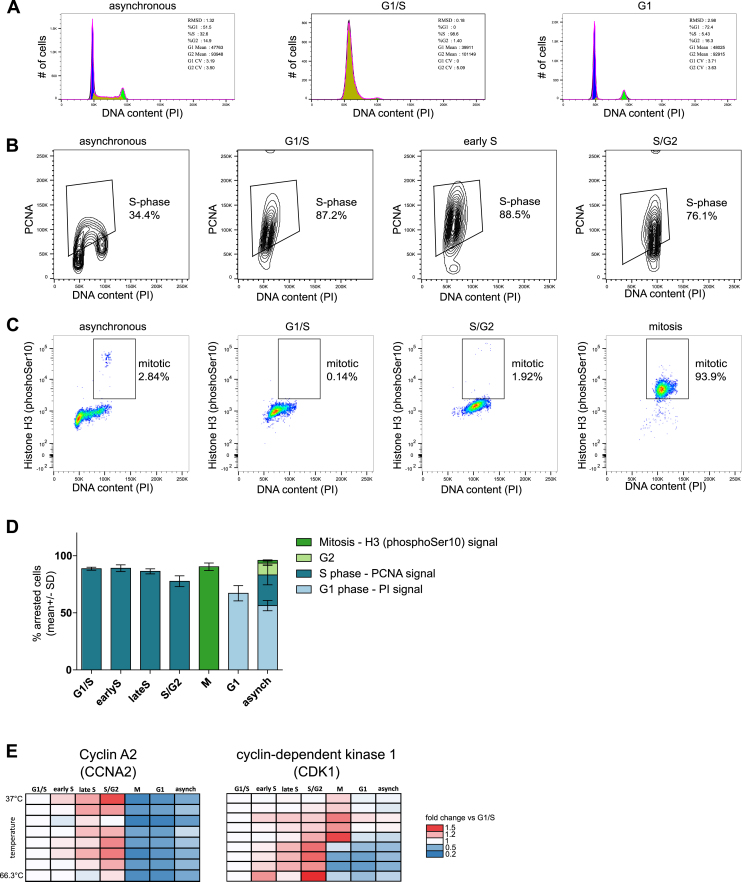
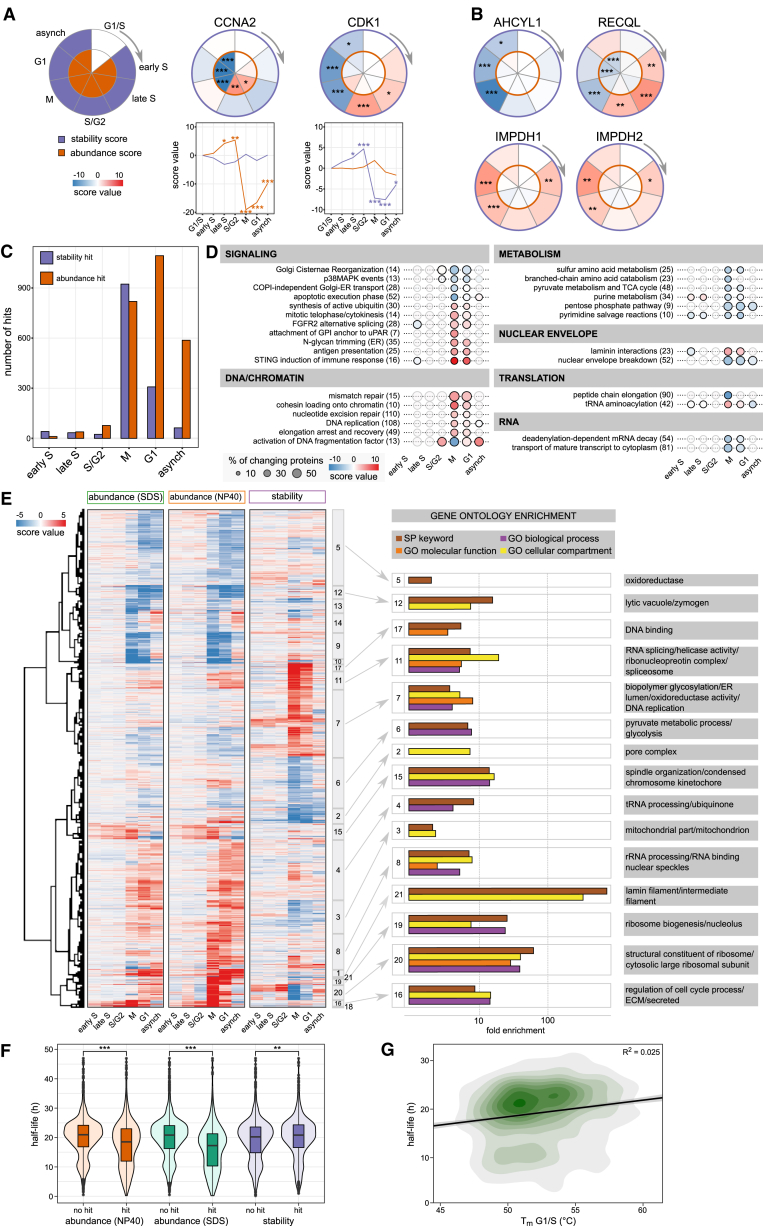
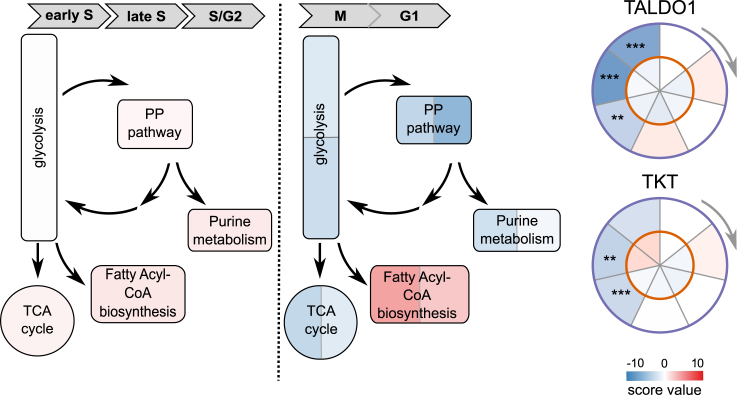
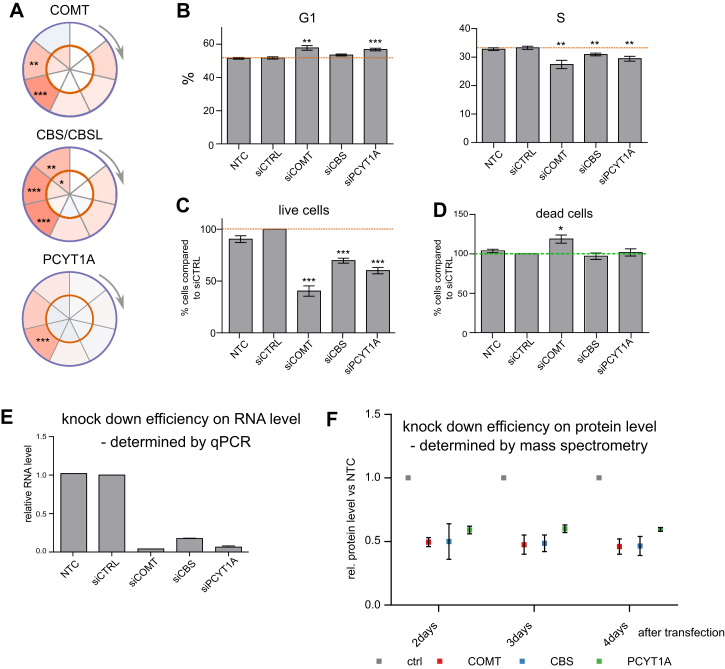
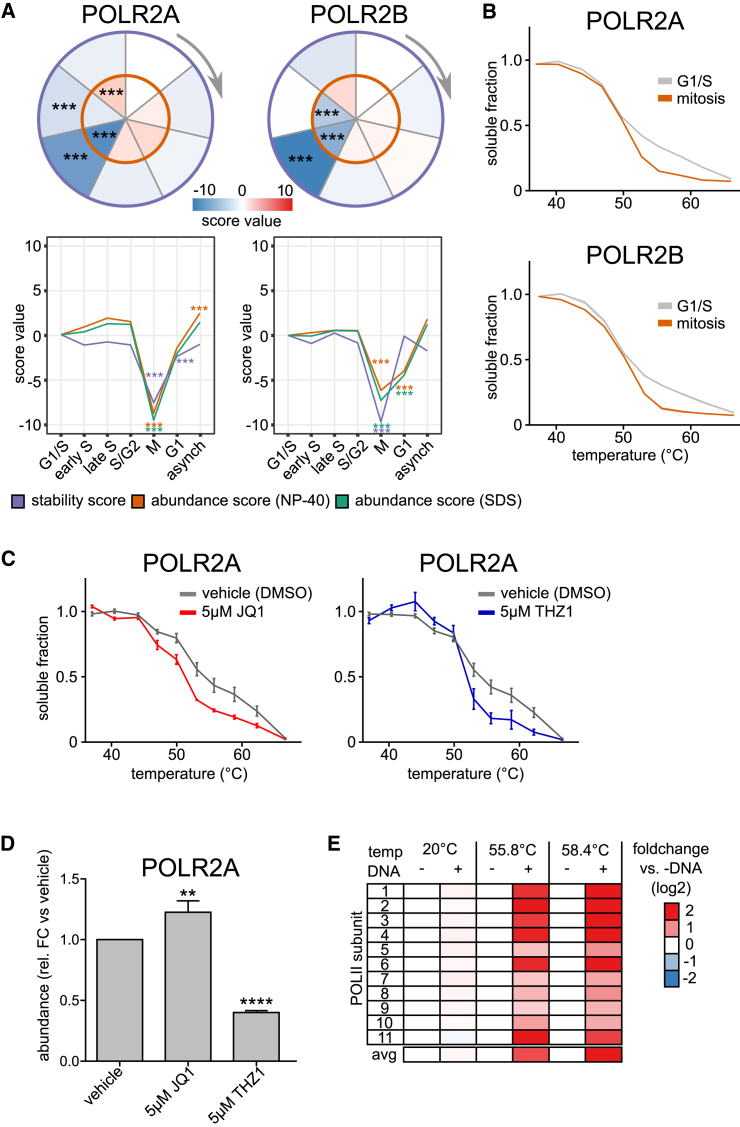
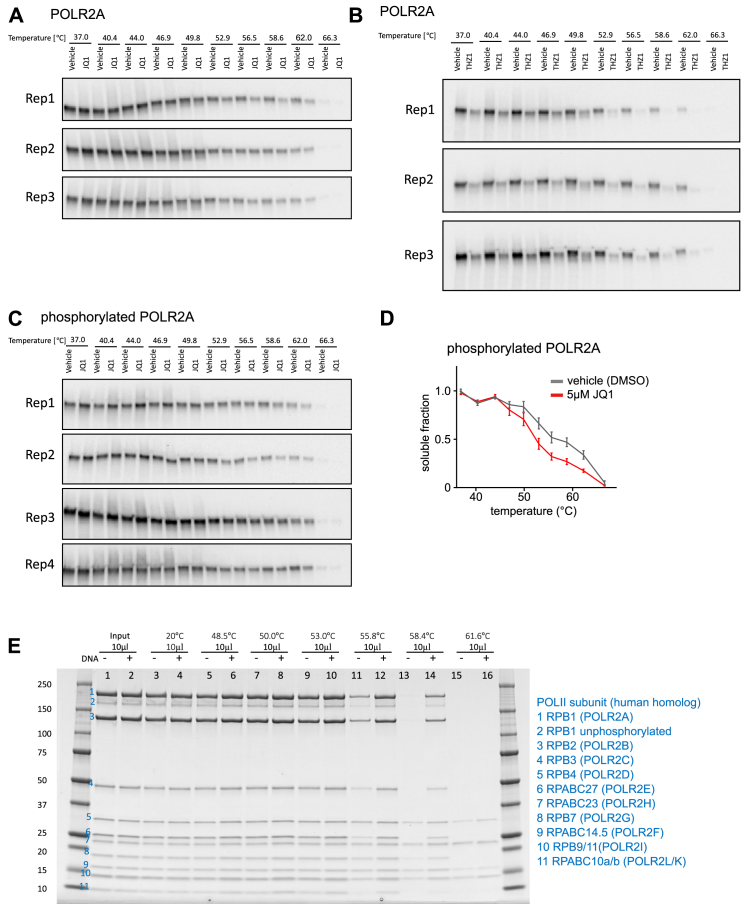
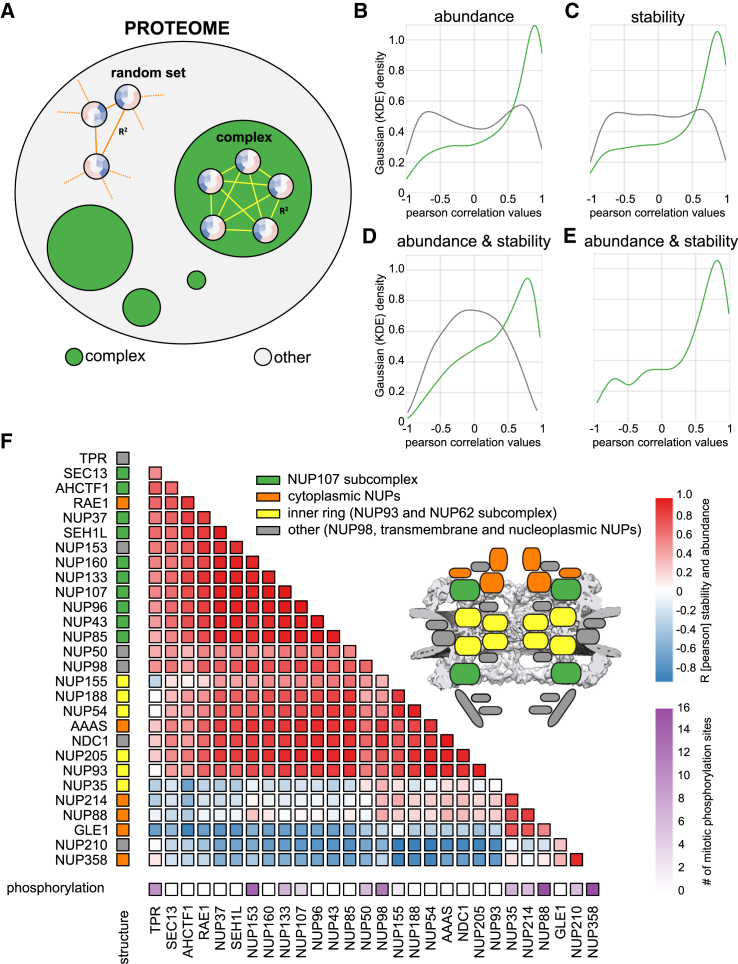
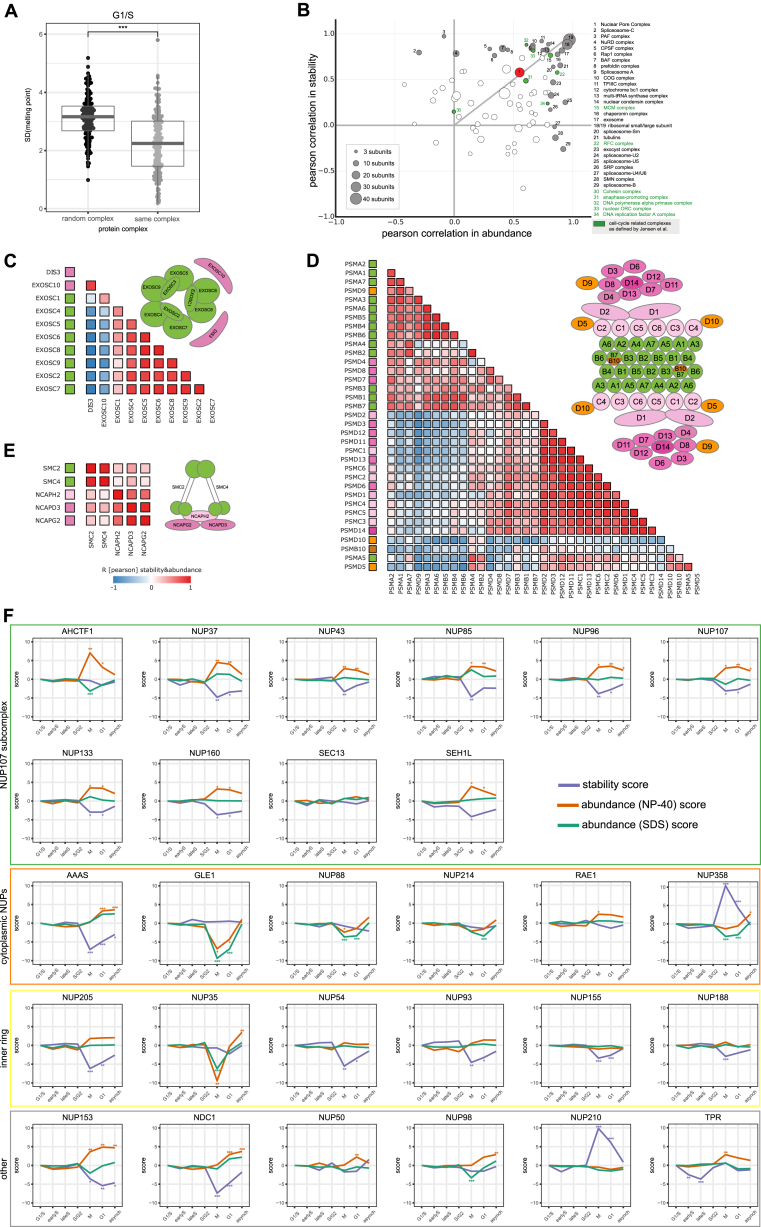
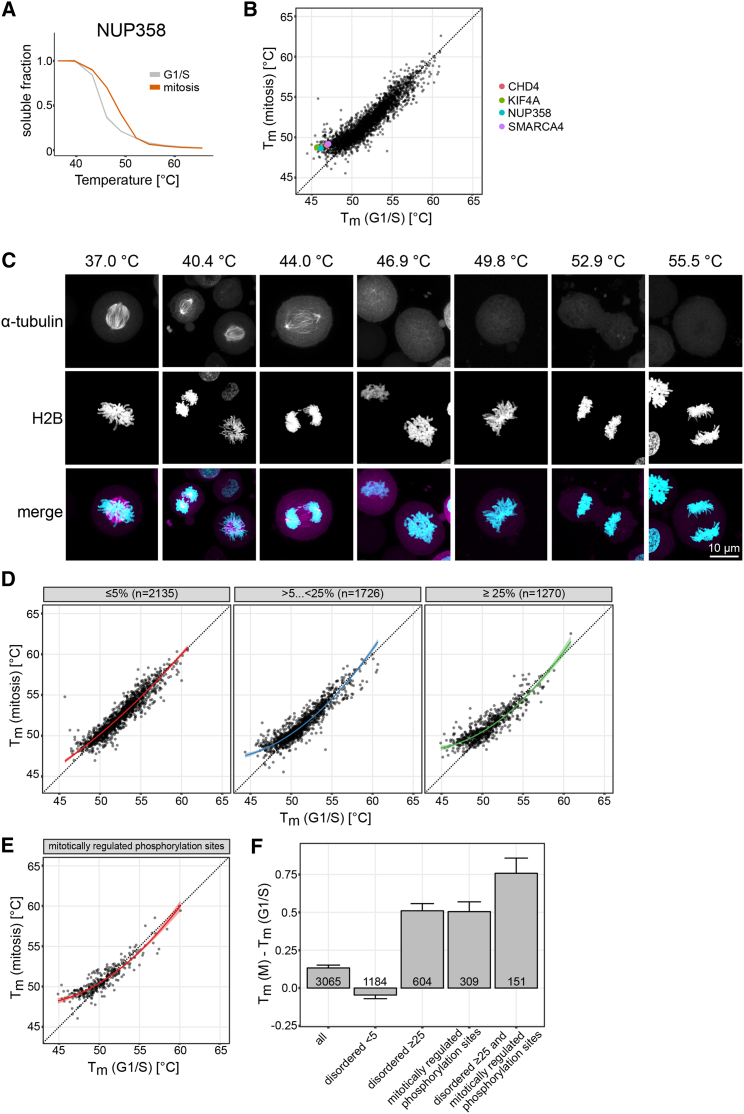
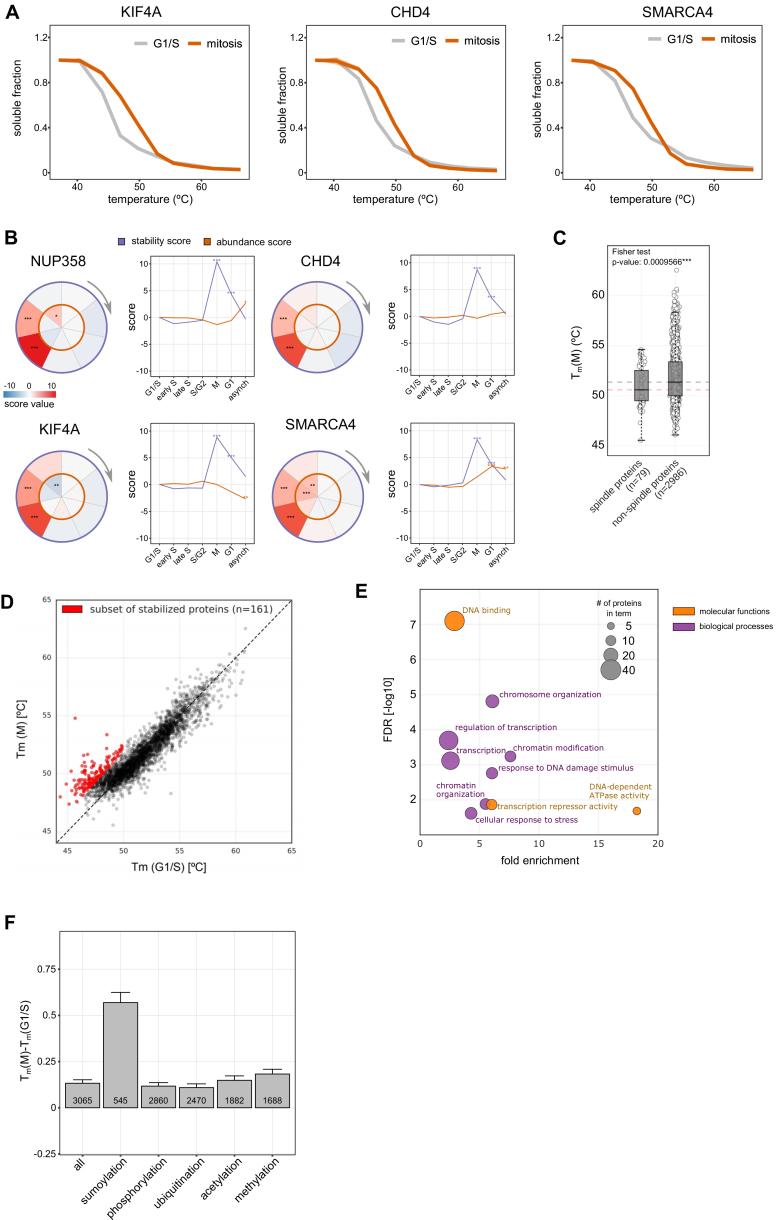
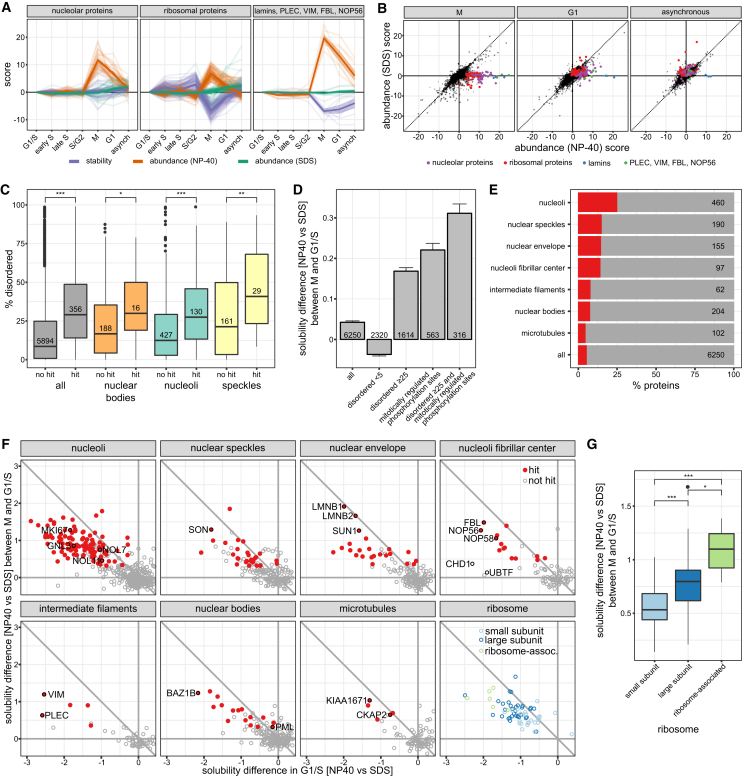
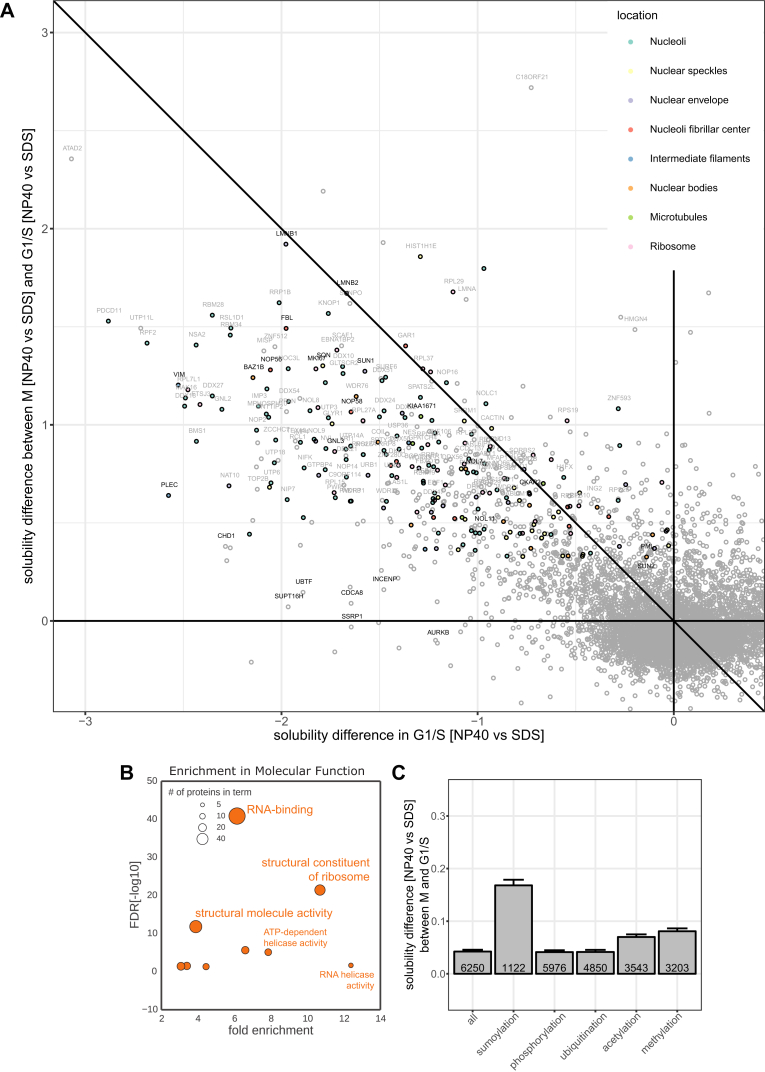
Quantitative mass spectrometry has established proteome-wide regulation of protein abundance and post-translational modifications in various biological processes. Here, we used quantitative mass spectrometry to systematically analyze the thermal stability and solubility of proteins on a proteome-wide scale during the eukaryotic cell cycle. We demonstrate pervasive variation of these biophysical parameters with most changes occurring in mitosis and G1. Various cellular pathways and components vary in thermal stability, such as cell-cycle factors, polymerases, and chromatin remodelers. We demonstrate that protein thermal stability serves as a proxy for enzyme activity, DNA binding, and complex formation in situ. Strikingly, a large cohort of intrinsically disordered and mitotically phosphorylated proteins is stabilized and solubilized in mitosis, suggesting a fundamental remodeling of the biophysical environment of the mitotic cell. Our data represent a rich resource for cell, structural, and systems biologists interested in proteome regulation during biological transitions.
Keywords: cell cycle; proteomics; thermal proteome profiling.
Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Bah A., Vernon R.M., Siddiqui Z., Krzeminski M., Muhandiram R., Zhao C., Sonenberg N., Kay L.E., Forman-Kay J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–109. - PubMed
-
- Becher I., Werner T., Doce C., Zaal E.A., Tögel I., Khan C.A., Rueger A., Muelbaier M., Salzer E., Berkers C.R. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 2016;12:908–910. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
