

Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation
- PMID: 29706549
- PMCID: PMC5989727
- DOI: 10.1016/j.cell.2018.03.074
Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation
Abstract
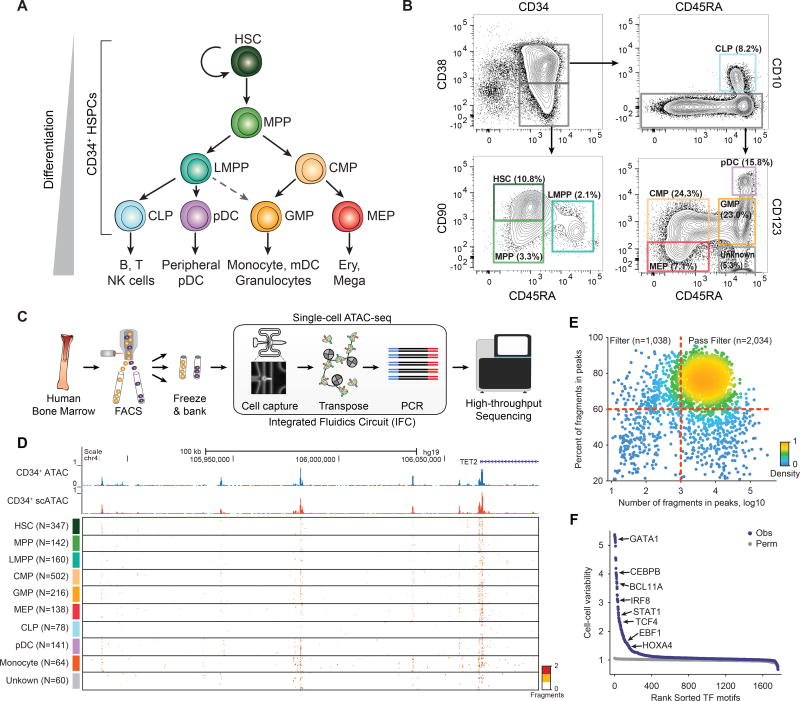
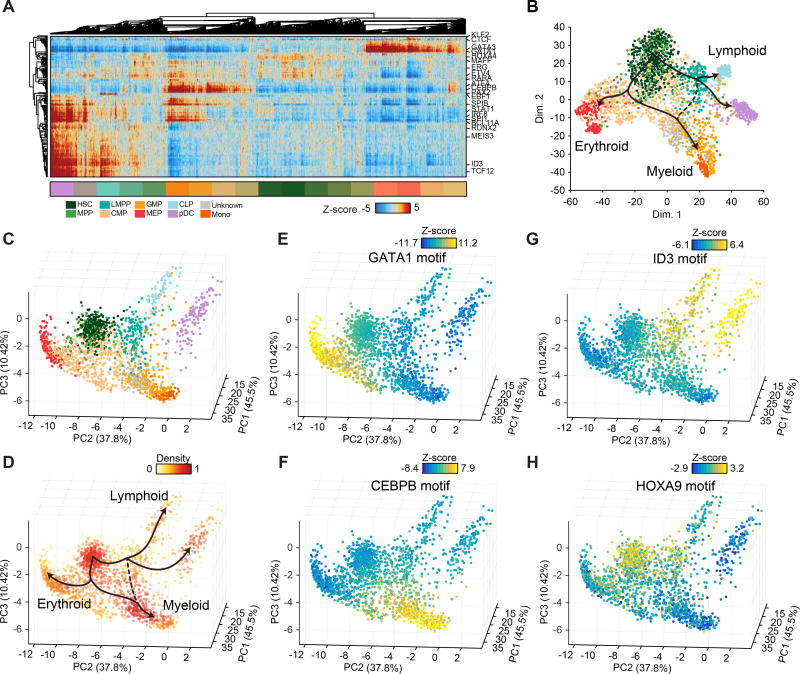
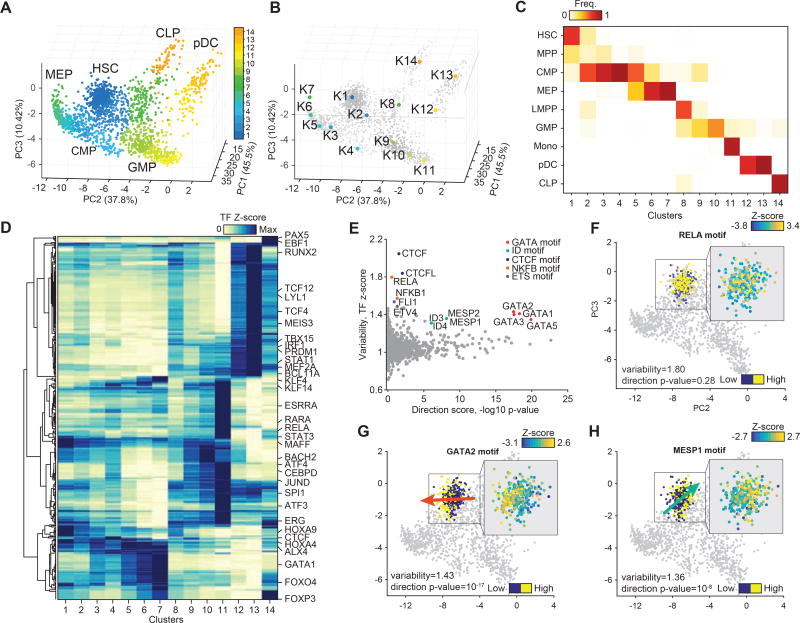
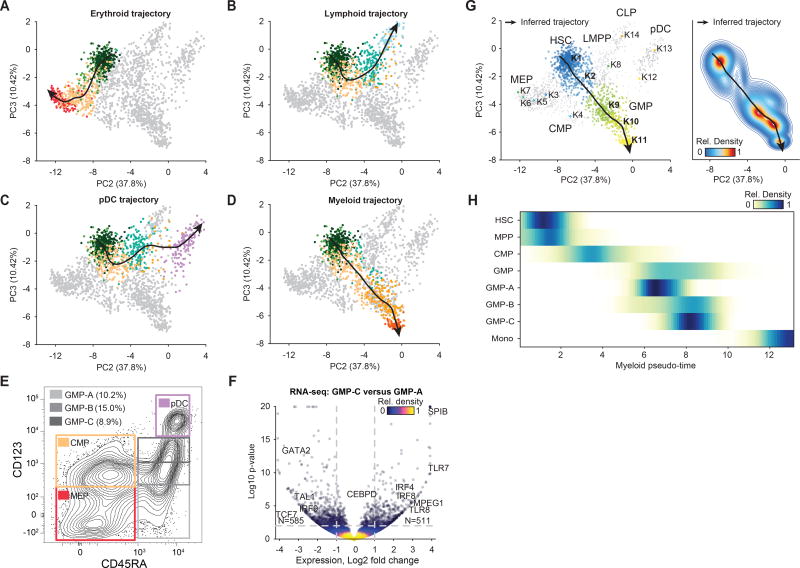
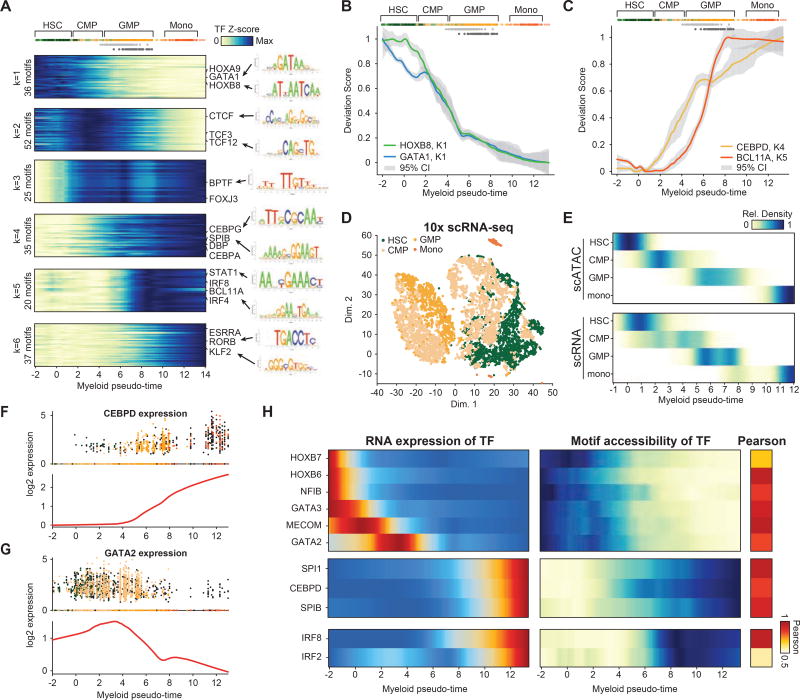
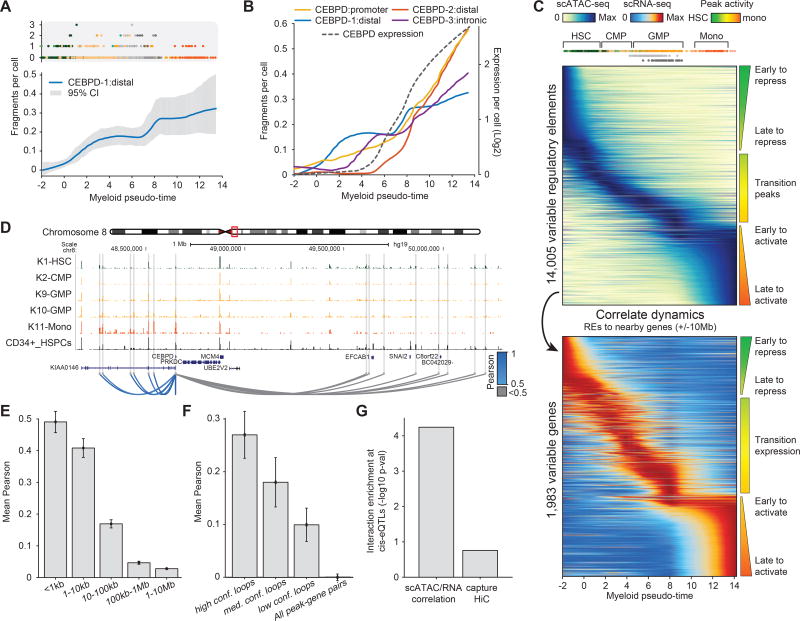
Human hematopoiesis involves cellular differentiation of multipotent cells into progressively more lineage-restricted states. While the chromatin accessibility landscape of this process has been explored in defined populations, single-cell regulatory variation has been hidden by ensemble averaging. We collected single-cell chromatin accessibility profiles across 10 populations of immunophenotypically defined human hematopoietic cell types and constructed a chromatin accessibility landscape of human hematopoiesis to characterize differentiation trajectories. We find variation consistent with lineage bias toward different developmental branches in multipotent cell types. We observe heterogeneity within common myeloid progenitors (CMPs) and granulocyte-macrophage progenitors (GMPs) and develop a strategy to partition GMPs along their differentiation trajectory. Furthermore, we integrated single-cell RNA sequencing (scRNA-seq) data to associate transcription factors to chromatin accessibility changes and regulatory elements to target genes through correlations of expression and regulatory element accessibility. Overall, this work provides a framework for integrative exploration of complex regulatory dynamics in a primary human tissue at single-cell resolution.
Keywords: chromatin accessibility; epigenomics; hematopoiesis; single cell.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. - PubMed
-
- Becker AJ, McCulloch EA, Till JE. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature. 1963;197:452–454. - PubMed
-
- Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. Accounting for technical noise in single-cell. RNA-seq experiments. 2013;10:1093–1095. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
