

Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy
- PMID: 29712858
- PMCID: PMC5960298
- DOI: 10.1073/pnas.1720065115
Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy
Abstract
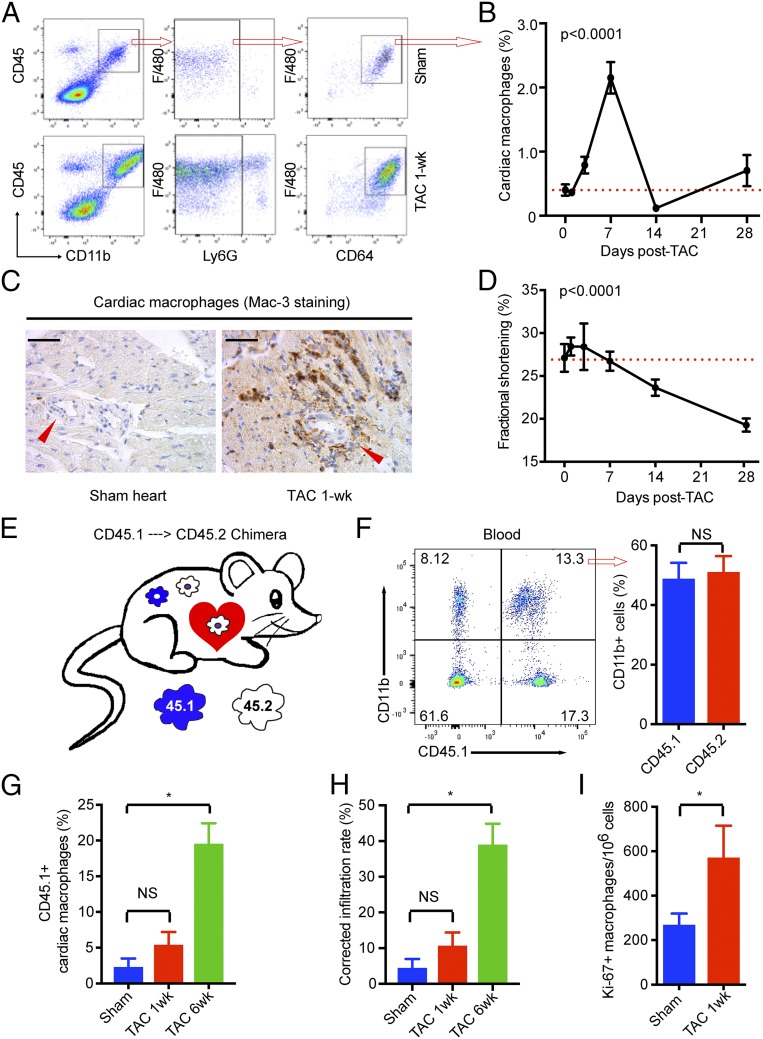
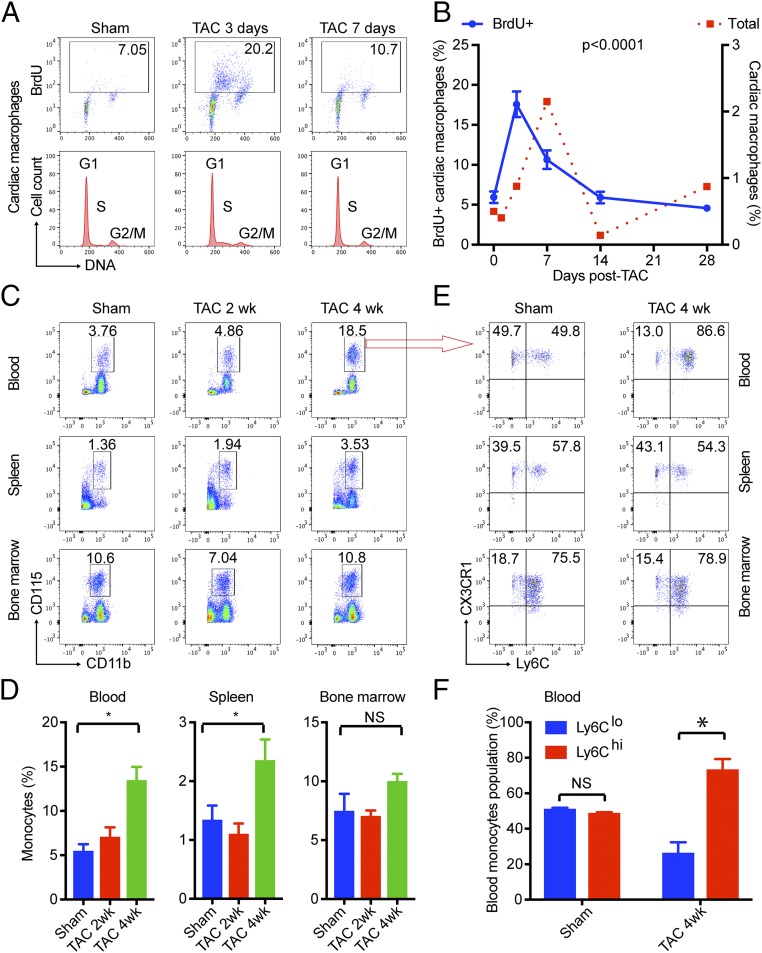
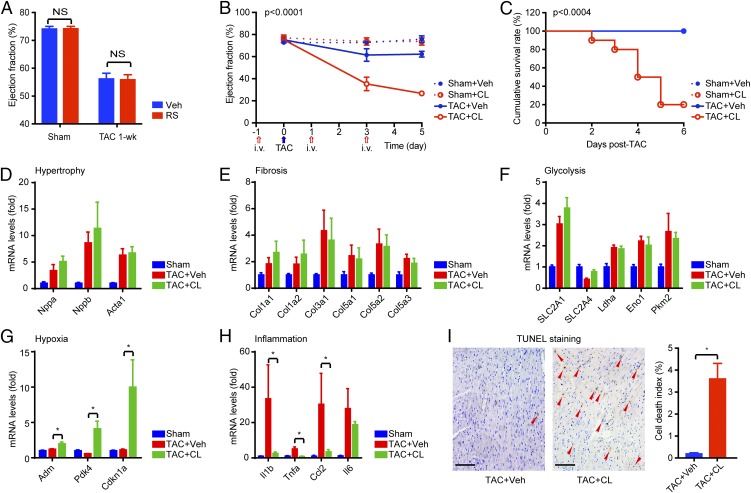
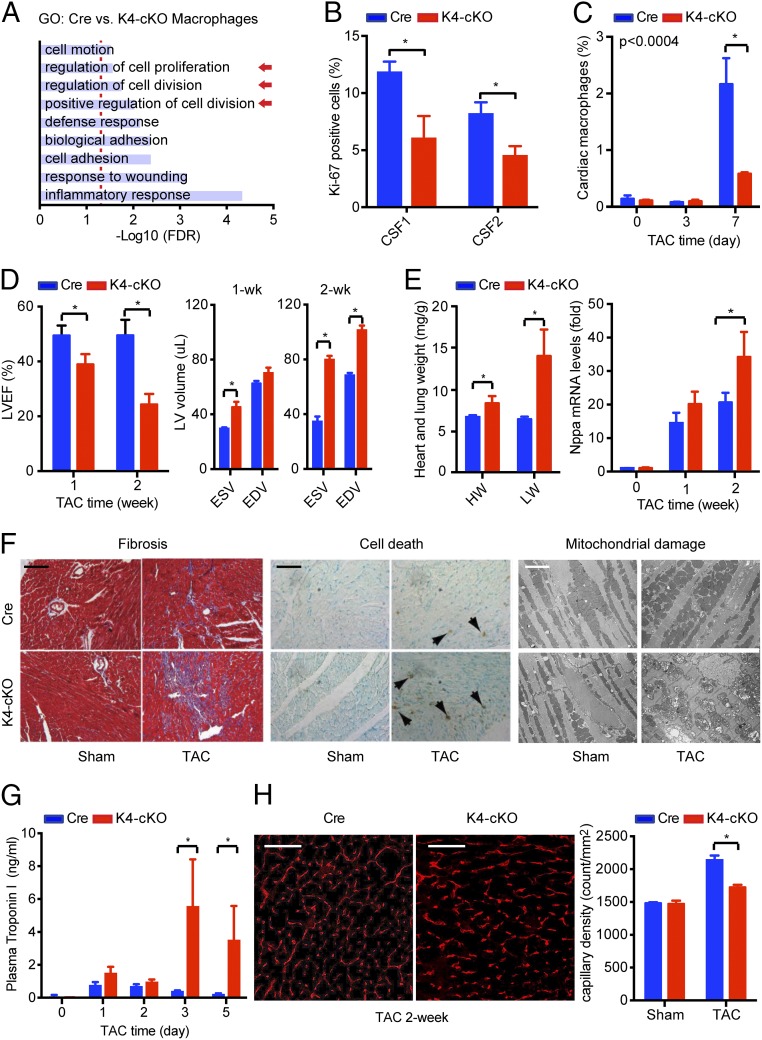
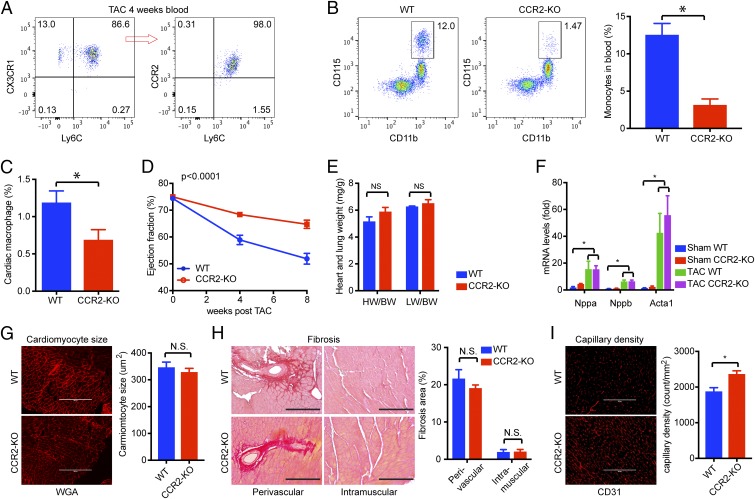
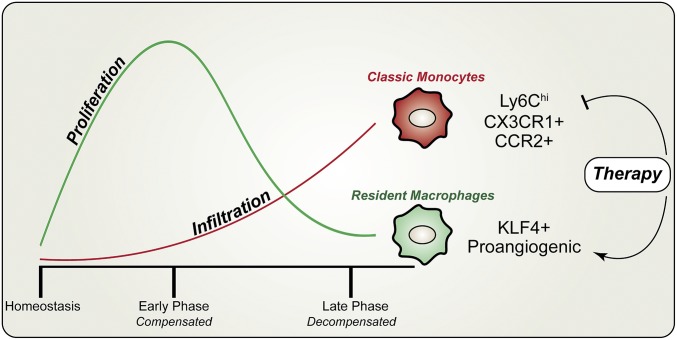
Nonischemic cardiomyopathy (NICM) resulting from long-standing hypertension, valvular disease, and genetic mutations is a major cause of heart failure worldwide. Recent observations suggest that myeloid cells can impact cardiac function, but the role of tissue-intrinsic vs. tissue-extrinsic myeloid cells in NICM remains poorly understood. Here, we show that cardiac resident macrophage proliferation occurs within the first week following pressure overload hypertrophy (POH; a model of heart failure) and is requisite for the heart's adaptive response. Mechanistically, we identify Kruppel-like factor 4 (KLF4) as a key transcription factor that regulates cardiac resident macrophage proliferation and angiogenic activities. Finally, we show that blood-borne macrophages recruited in late-phase POH are detrimental, and that blockade of their infiltration improves myocardial angiogenesis and preserves cardiac function. These observations demonstrate previously unappreciated temporal and spatial roles for resident and nonresident macrophages in the development of heart failure.
Keywords: angiogenesis; cardiac macrophage; pressure overload hypertrophy.
Conflict of interest statement
Conflict of interest statement: X.L. and M.K.J. have a patent application (serial no. 62/644,792).
Figures
References
-
- Cecchi F, et al. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349:1027–1035. - PubMed
-
- Treasure CB, et al. Hypertension and left ventricular hypertrophy are associated with impaired endothelium-mediated relaxation in human coronary resistance vessels. Circulation. 1993;87:86–93. - PubMed
-
- Maron BJ, Wolfson JK, Epstein SE, Roberts WC. Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1986;8:545–557. - PubMed
-
- Frey N, Luedde M, Katus HA. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat Rev Cardiol. 2011;9:91–100. - PubMed
-
- Sano M, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
