

Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes
- PMID: 29713087
- PMCID: PMC6613387
- DOI: 10.1038/s41591-018-0016-8
Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes
Erratum in
-
Publisher Correction: Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes.Nat Med. 2018 Aug;24(8):1292. doi: 10.1038/s41591-018-0098-3. Nat Med. 2018. PMID: 29955181
-
Author Correction: Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes.Nat Med. 2018 Aug;24(8):1290-1291. doi: 10.1038/s41591-018-0097-4. Nat Med. 2018. PMID: 29955182
Abstract
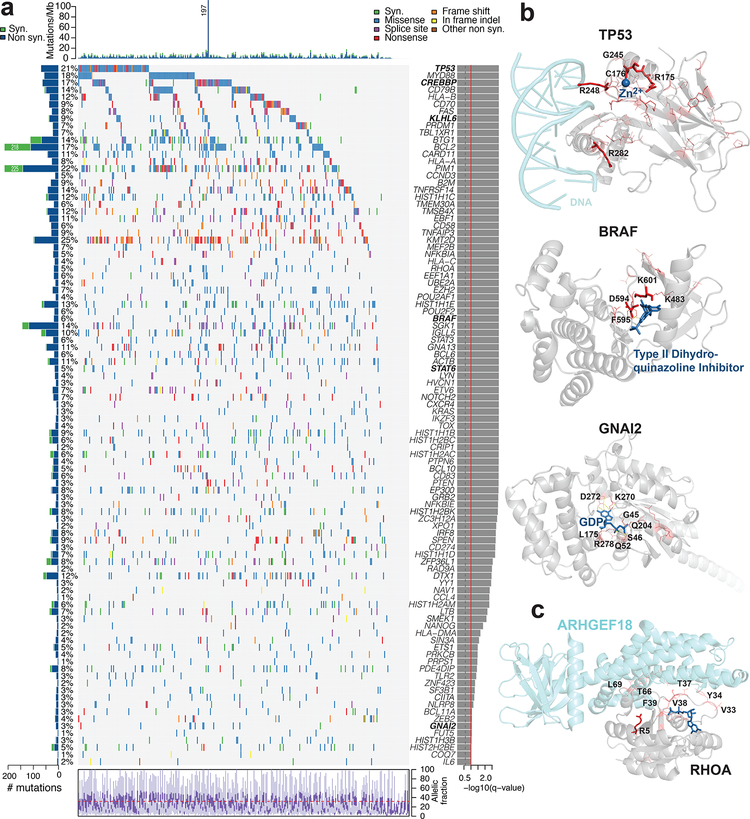
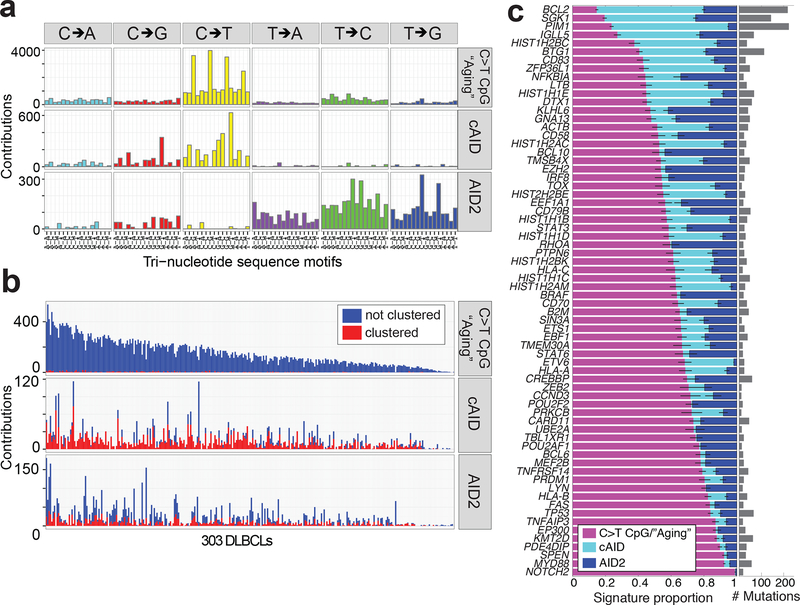
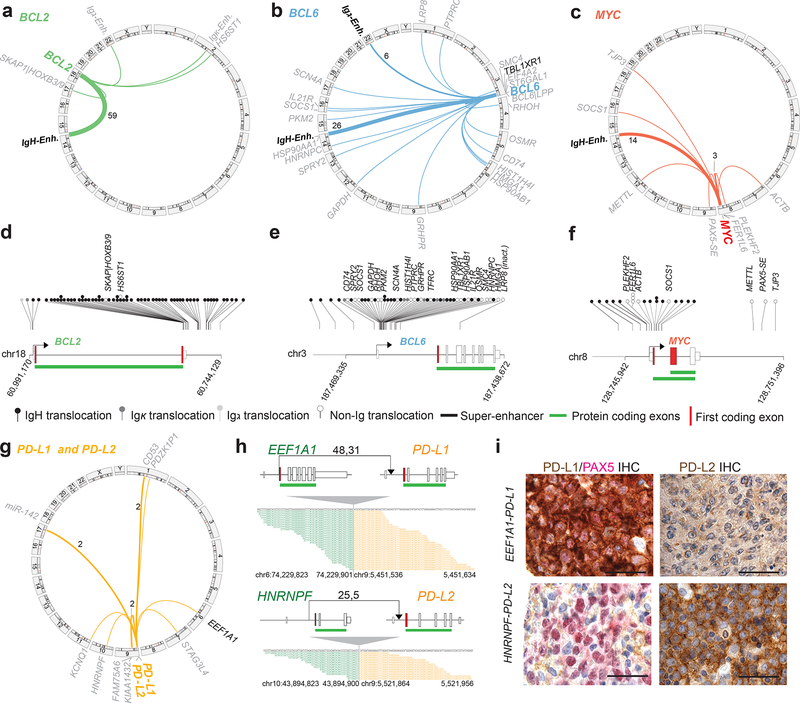
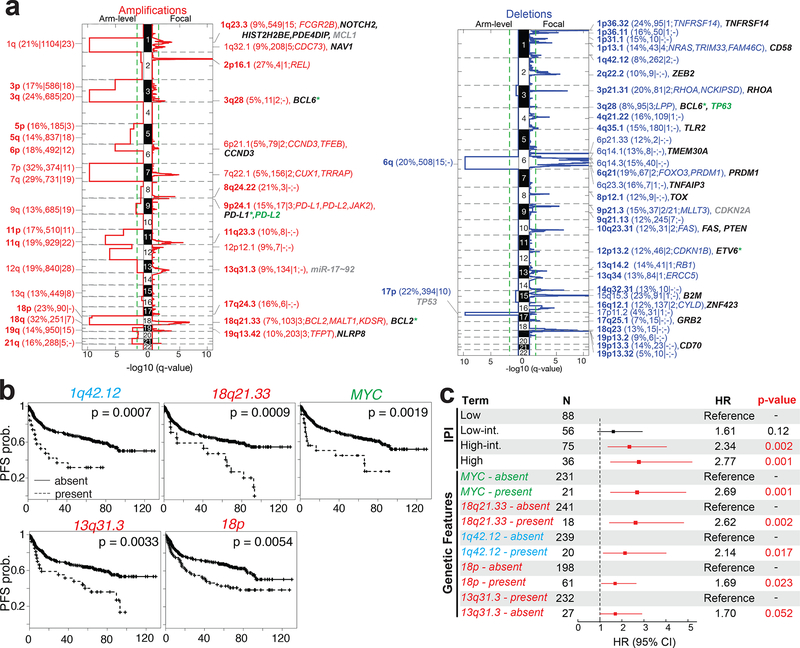
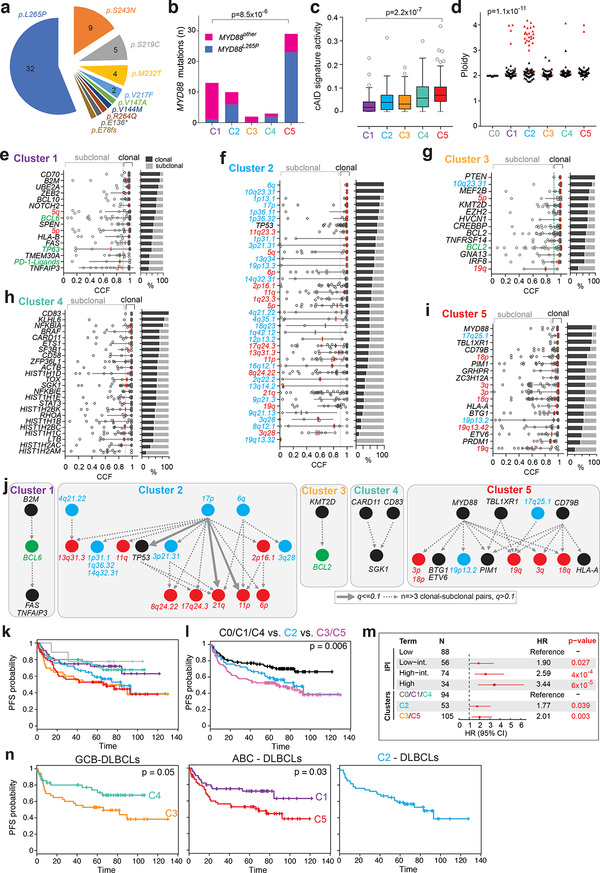
Diffuse large B cell lymphoma (DLBCL), the most common lymphoid malignancy in adults, is a clinically and genetically heterogeneous disease that is further classified into transcriptionally defined activated B cell (ABC) and germinal center B cell (GCB) subtypes. We carried out a comprehensive genetic analysis of 304 primary DLBCLs and identified low-frequency alterations, captured recurrent mutations, somatic copy number alterations, and structural variants, and defined coordinate signatures in patients with available outcome data. We integrated these genetic drivers using consensus clustering and identified five robust DLBCL subsets, including a previously unrecognized group of low-risk ABC-DLBCLs of extrafollicular/marginal zone origin; two distinct subsets of GCB-DLBCLs with different outcomes and targetable alterations; and an ABC/GCB-independent group with biallelic inactivation of TP53, CDKN2A loss, and associated genomic instability. The genetic features of the newly characterized subsets, their mutational signatures, and the temporal ordering of identified alterations provide new insights into DLBCL pathogenesis. The coordinate genetic signatures also predict outcome independent of the clinical International Prognostic Index and suggest new combination treatment strategies. More broadly, our results provide a roadmap for an actionable DLBCL classification.
Conflict of interest statement
Competing Financial Interest Statement
The authors declare that they have no competing financial interests.
Figures

References
-
- Basso K & Dalla-Favera R Germinal centres and B cell lymphomagenesis. Nat Rev Immunol 15, 172–184 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
