

A genome-wide assessment of conserved SNP alleles reveals a panel of regulatory SNPs relevant to the peripheral nerve
- PMID: 29716548
- PMCID: PMC5930951
- DOI: 10.1186/s12864-018-4692-z
A genome-wide assessment of conserved SNP alleles reveals a panel of regulatory SNPs relevant to the peripheral nerve
Abstract
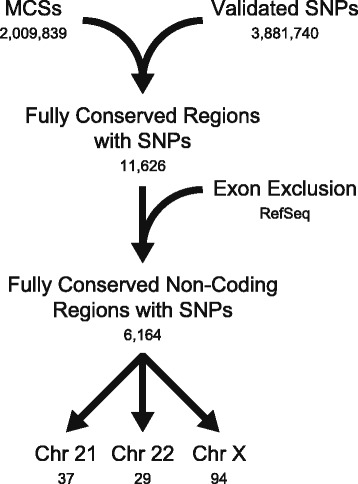
Background: Identifying functional non-coding variation is critical for defining the genetic contributions to human disease. While single-nucleotide polymorphisms (SNPs) within cis-acting transcriptional regulatory elements have been implicated in disease pathogenesis, not all cell types have been assessed and functional validations have been limited. In particular, the cells of the peripheral nervous system have been excluded from genome-wide efforts to link non-coding SNPs to altered gene function. Addressing this gap is essential for defining the genetic architecture of diseases that affect the peripheral nerve. We developed a computational pipeline to identify SNPs that affect regulatory function (rSNPs) and evaluated our predictions on a set of 144 regions in Schwann cells, motor neurons, and muscle cells.
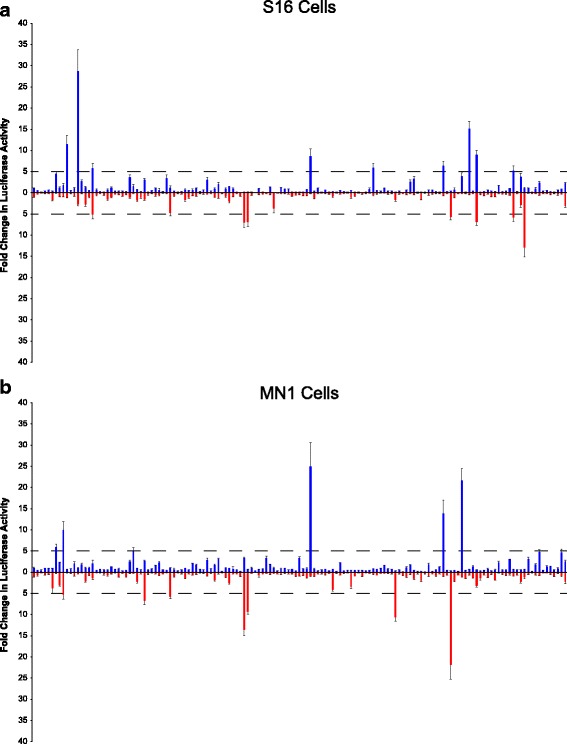
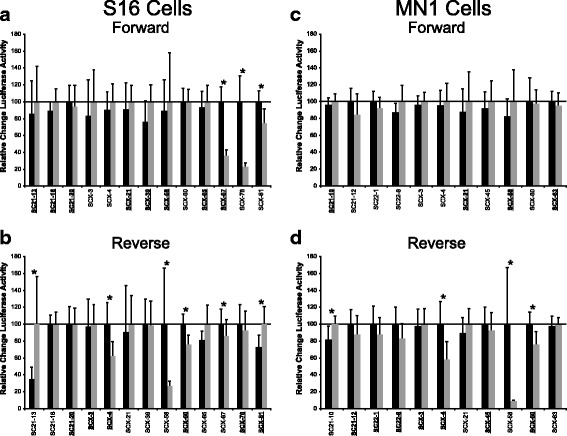
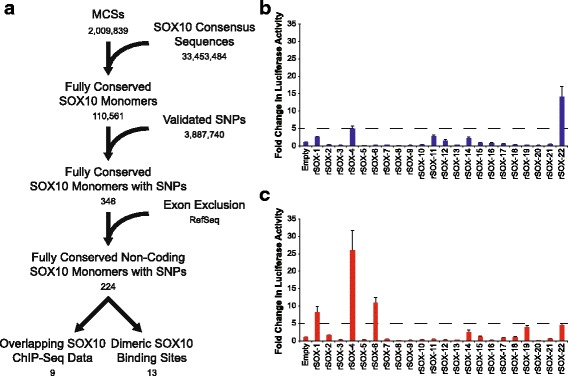
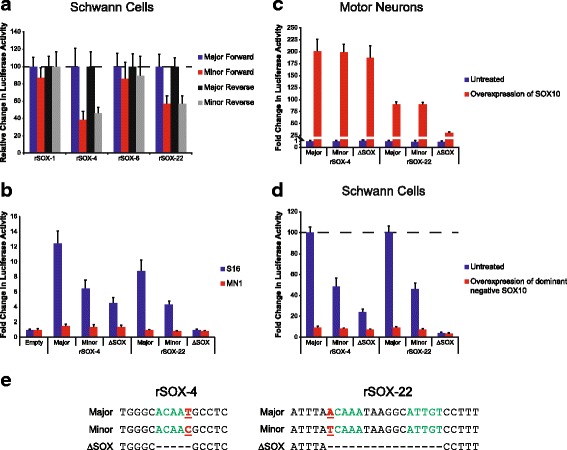
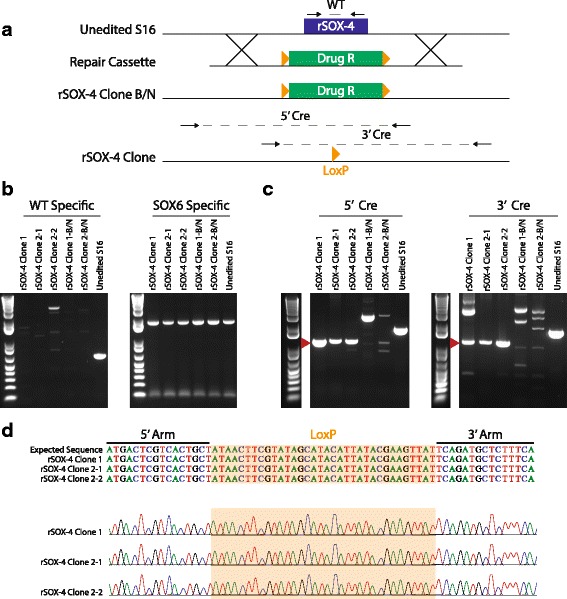
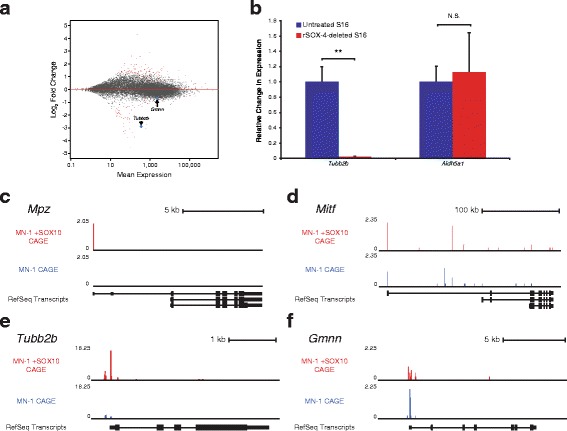
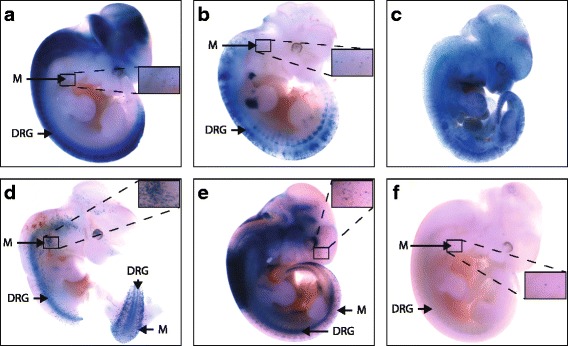
Results: We identified 28 regions that display regulatory activity in at least one cell type and 13 SNPs that affect regulatory function. We then tailored our pipeline to one peripheral nerve cell type by incorporating SOX10 ChIP-Seq data; SOX10 is essential for Schwann cells. We prioritized 22 putative SOX10 response elements harboring a SNP and rapidly validated two rSNPs. We then selected one of these elements for further characterization to assess the biological relevance of our approach. Deletion of the element from the genome of cultured Schwann cells-followed by differential gene expression studies-revealed Tubb2b as a candidate target gene. Studying the enhancer in developing mouse embryos revealed activity in SOX10-positive cells including the dorsal root ganglia and melanoblasts.
Conclusions: Our efforts provide insight into the utility of employing strict conservation for rSNP discovery. This strategy, combined with functional analyses, can yield candidate target genes. In support of this, our efforts suggest that investigating the role of Tubb2b in SOX10-positive cells may reveal novel biology within these cell populations.
Keywords: Enhancer; Neuropathy; Peripheral nerve; SOX10; Schwann cells; TUBB2B; Transcriptional regulation.
Conflict of interest statement
Ethics approval and consent to participate
The mouse transgenic experiments were performed by the University of Michigan Transgenic Animal Core and were approved by the University of Michigan Committee on the Use and Care of Animals.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12(14):1725–1735. doi: 10.1093/hmg/ddg180. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
