

Genome-wide Analysis of Large-scale Longitudinal Outcomes using Penalization -GALLOP algorithm
- PMID: 29717146
- PMCID: PMC5931565
- DOI: 10.1038/s41598-018-24578-7
Genome-wide Analysis of Large-scale Longitudinal Outcomes using Penalization -GALLOP algorithm
Abstract
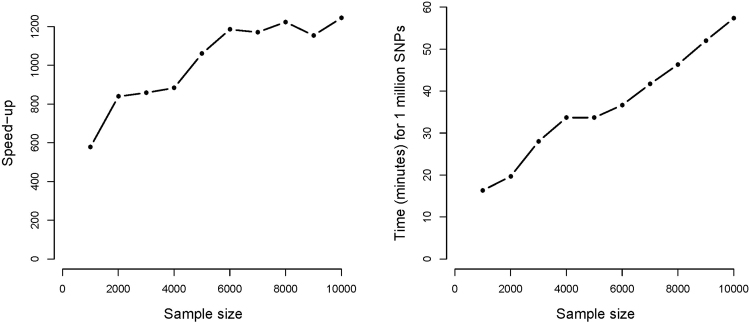
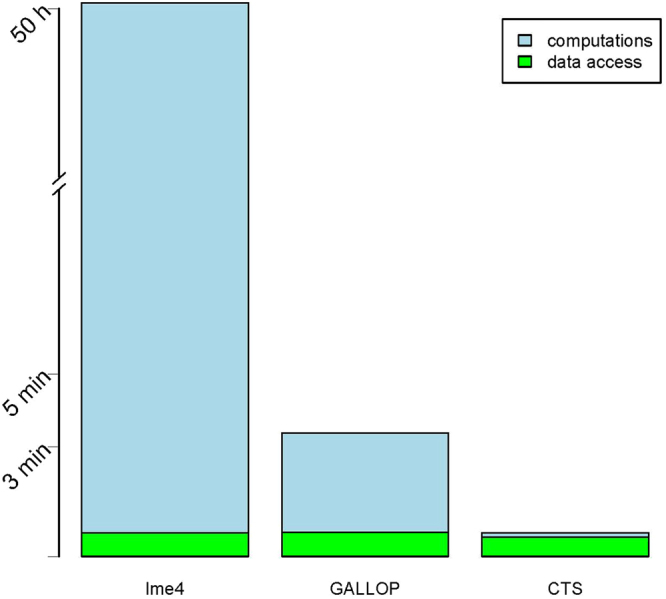
Genome-wide association studies (GWAS) with longitudinal phenotypes provide opportunities to identify genetic variations associated with changes in human traits over time. Mixed models are used to correct for the correlated nature of longitudinal data. GWA studies are notorious for their computational challenges, which are considerable when mixed models for thousands of individuals are fitted to millions of SNPs. We present a new algorithm that speeds up a genome-wide analysis of longitudinal data by several orders of magnitude. It solves the equivalent penalized least squares problem efficiently, computing variances in an initial step. Factorizations and transformations are used to avoid inversion of large matrices. Because the system of equations is bordered, we can re-use components, which can be precomputed for the mixed model without a SNP. Two SNP effects (main and its interaction with time) are obtained. Our method completes the analysis a thousand times faster than the R package lme4, providing an almost identical solution for the coefficients and p-values. We provide an R implementation of our algorithm.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
