

Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations
- PMID: 29719671
- PMCID: PMC5921983
- DOI: 10.1186/s13229-018-0205-9
Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations
Abstract
Background: Phelan-McDermid syndrome (PMS) is a neurodevelopmental disorder characterized by psychiatric and neurological features. Most reported cases are caused by 22q13.3 deletions, leading to SHANK3 haploinsufficiency, but also usually encompassing many other genes. While the number of point mutations identified in SHANK3 has increased in recent years due to large-scale sequencing studies, systematic studies describing the phenotype of individuals harboring such mutations are lacking.
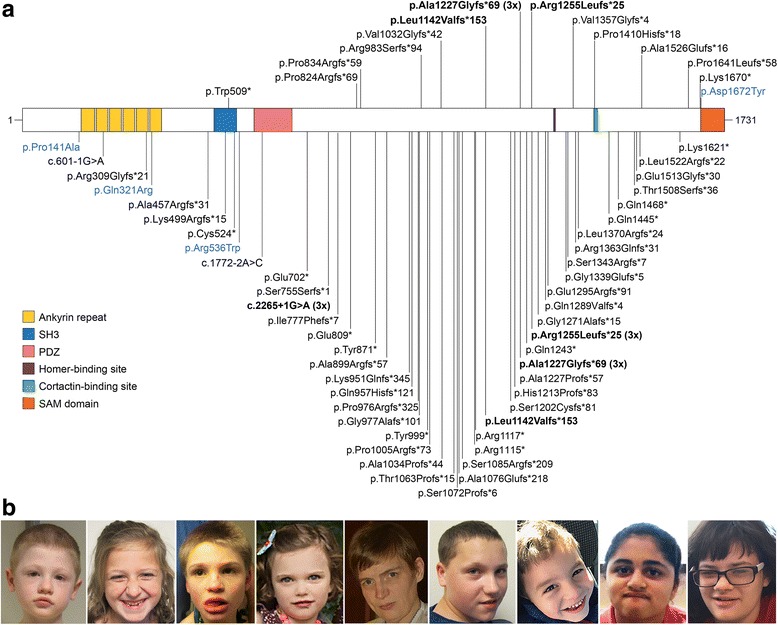
Methods: We provide detailed clinical and genetic data on 17 individuals carrying mutations in SHANK3. We also review 60 previously reported patients with pathogenic or likely pathogenic SHANK3 variants, often lacking detailed phenotypic information.
Results: SHANK3 mutations in our cohort and in previously reported cases were distributed throughout the protein; the majority were truncating and all were compatible with de novo inheritance. Despite substantial allelic heterogeneity, four variants were recurrent (p.Leu1142Valfs*153, p.Ala1227Glyfs*69, p.Arg1255Leufs*25, and c.2265+1G>A), suggesting that these are hotspots for de novo mutations. All individuals studied had intellectual disability, and autism spectrum disorder was prevalent (73%). Severe speech deficits were common, but in contrast to individuals with 22q13.3 deletions, the majority developed single words, including 41% with at least phrase speech. Other common findings were consistent with reports among individuals with 22q13.3 deletions, including hypotonia, motor skill deficits, regression, seizures, brain abnormalities, mild dysmorphic features, and feeding and gastrointestinal problems.
Conclusions: Haploinsufficiency of SHANK3 resulting from point mutations is sufficient to cause a broad range of features associated with PMS. Our findings expand the molecular and phenotypic spectrum of PMS caused by SHANK3 point mutations and suggest that, in general, speech impairment and motor deficits are more severe in the case of deletions. In contrast, renal abnormalities associated with 22q13.3 deletions do not appear to be related to the loss of SHANK3.
Keywords: 22q13 deletion syndrome; Autism spectrum disorder; Intellectual disability; Phelan-McDermid syndrome; Phenotype; SHANK3; Sequence variants.
Conflict of interest statement
The institutional review boards of the Icahn School of Medicine at Mount Sinai and Baylor College of Medicine approved this study. Participants were enrolled after written informed consent was obtained from parents or legal guardians.Written informed consent for publication was obtained from the parents or legal guardians.JDB and Mount Sinai hold a shared parent for the use of insulin-like growth factor-1 (IGF-1) in Phelan-McDermid syndrome. JDB is on the scientific advisory board for Coronis Neuroscience and has consulted for the Gerson Lehrman Group. AK is on the advisory board of Vencerx Therapeutics and consults for Ovid Therapeutics. The remaining authors declare that they have no competing interests.Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M, et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observations. J Med Genet. 2003;40:690–696. doi: 10.1136/jmg.40.9.690. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
