

SHI7 Is a Self-Learning Pipeline for Multipurpose Short-Read DNA Quality Control
- PMID: 29719872
- PMCID: PMC5915699
- DOI: 10.1128/mSystems.00202-17
SHI7 Is a Self-Learning Pipeline for Multipurpose Short-Read DNA Quality Control
Abstract
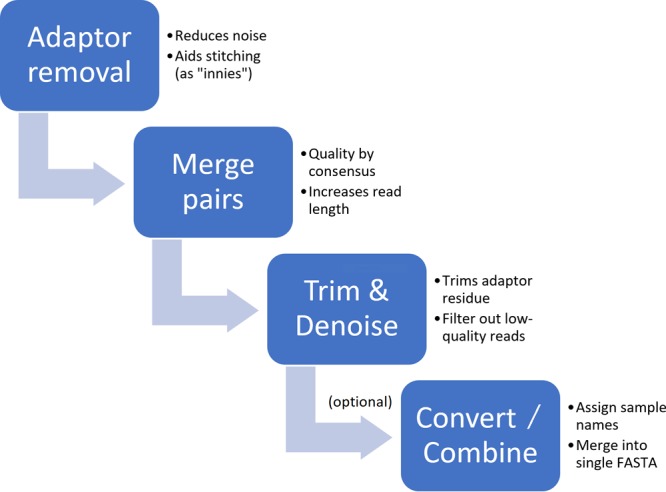
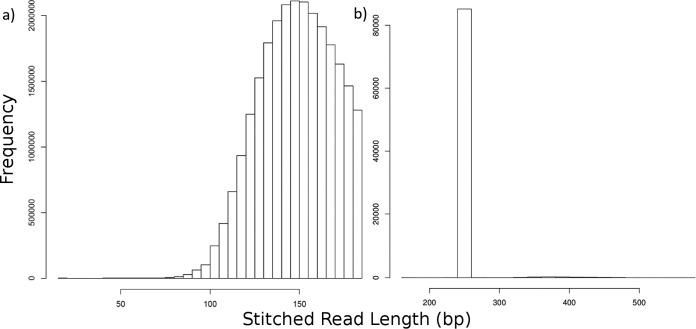
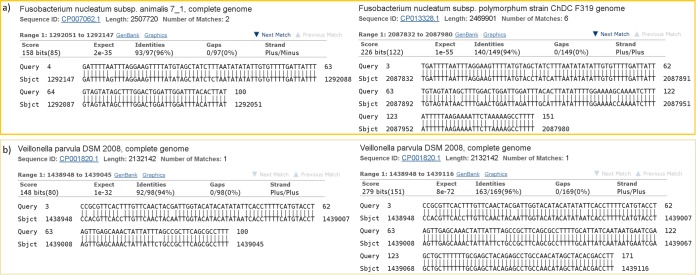
Next-generation sequencing technology is of great importance for many biological disciplines; however, due to technical and biological limitations, the short DNA sequences produced by modern sequencers require numerous quality control (QC) measures to reduce errors, remove technical contaminants, or merge paired-end reads together into longer or higher-quality contigs. Many tools for each step exist, but choosing the appropriate methods and usage parameters can be challenging because the parameterization of each step depends on the particularities of the sequencing technology used, the type of samples being analyzed, and the stochasticity of the instrumentation and sample preparation. Furthermore, end users may not know all of the relevant information about how their data were generated, such as the expected overlap for paired-end sequences or type of adaptors used to make informed choices. This increasing complexity and nuance demand a pipeline that combines existing steps together in a user-friendly way and, when possible, learns reasonable quality parameters from the data automatically. We propose a user-friendly quality control pipeline called SHI7 (canonically pronounced "shizen"), which aims to simplify quality control of short-read data for the end user by predicting presence and/or type of common sequencing adaptors, what quality scores to trim, whether the data set is shotgun or amplicon sequencing, whether reads are paired end or single end, and whether pairs are stitchable, including the expected amount of pair overlap. We hope that SHI7 will make it easier for all researchers, expert and novice alike, to follow reasonable practices for short-read data quality control. IMPORTANCE Quality control of high-throughput DNA sequencing data is an important but sometimes laborious task requiring background knowledge of the sequencing protocol used (such as adaptor type, sequencing technology, insert size/stitchability, paired-endedness, etc.). Quality control protocols typically require applying this background knowledge to selecting and executing numerous quality control steps with the appropriate parameters, which is especially difficult when working with public data or data from collaborators who use different protocols. We have created a streamlined quality control pipeline intended to substantially simplify the process of DNA quality control from raw machine output files to actionable sequence data. In contrast to other methods, our proposed pipeline is easy to install and use and attempts to learn the necessary parameters from the data automatically with a single command.
Keywords: QC; algorithm; bioinformatics; metagenomics; microbiome; pipeline; quality control; sequencing; short read.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
