

GNE myopathy: from clinics and genetics to pathology and research strategies
- PMID: 29720219
- PMCID: PMC5930817
- DOI: 10.1186/s13023-018-0802-x
GNE myopathy: from clinics and genetics to pathology and research strategies
Abstract
GNE myopathy is an ultra-rare autosomal recessive disease, which starts as a distal muscle weakness and ultimately leads to a wheelchair bound state. Molecular research and animal modelling significantly moved forward understanding of GNE myopathy mechanisms and suggested therapeutic interventions to alleviate the symptoms. Multiple therapeutic attempts are being made to supplement sialic acid depleted in GNE myopathy muscle cells. Translational research field provided valuable knowledge through natural history studies, patient registries and clinical trial, which significantly contributed to bringing forward an era of GNE myopathy treatment. In this review, we are summarising current GNE myopathy, scientific trends and open questions, which would be of significant interest for a wide neuromuscular diseases community.
Keywords: DMRV; Distal myopathy; GNE myopathy; HIBM; Nonaka disease; QSM; Sialic acid.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
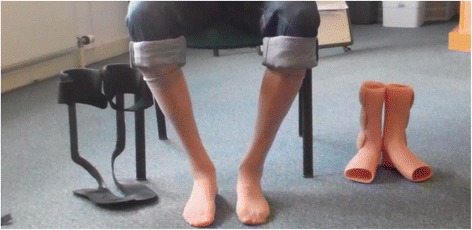
The authors have consent to use the image shown in Fig. 1.
Competing interests
The authors declare that they do not have any competing interests.
OP and HL have served as investigators on clinical trials and and a Disease Monitoring Programme for GNE myopathy carried out by Newcastle University and the Newcastle NHS Trust sponsored by Ultragenyx Pharmaceuticals (NCT01784679, NCT02377921, NCT02736188).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
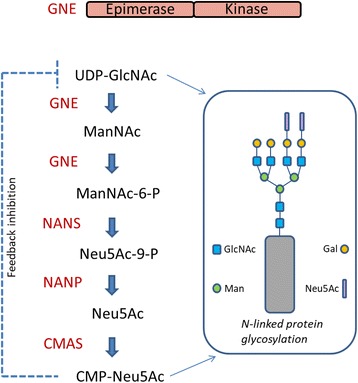
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–87. doi: 10.1038/ng718. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
