

Efficient exon skipping of SGCG mutations mediated by phosphorodiamidate morpholino oligomers
- PMID: 29720576
- PMCID: PMC6012523
- DOI: 10.1172/jci.insight.99357
Efficient exon skipping of SGCG mutations mediated by phosphorodiamidate morpholino oligomers
Abstract
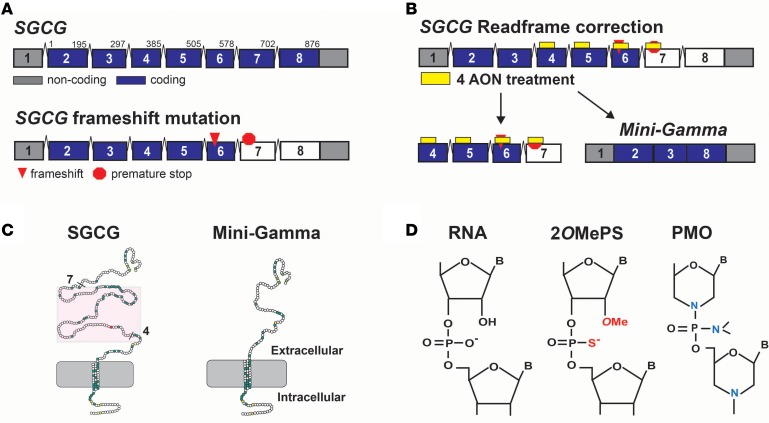
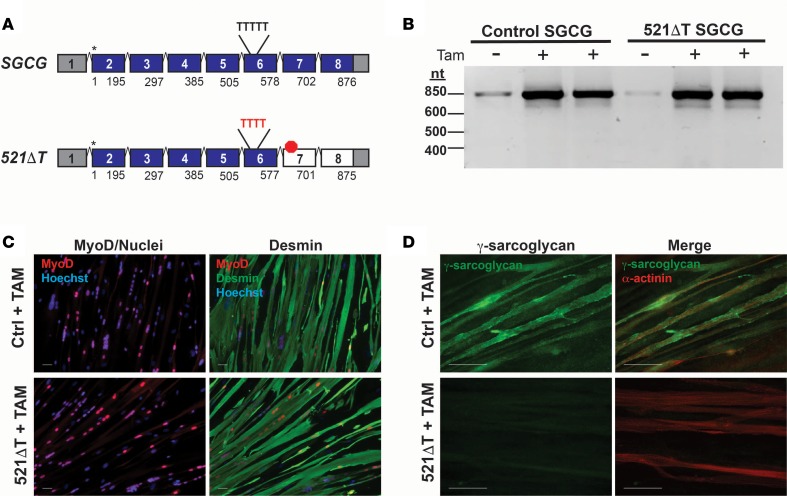
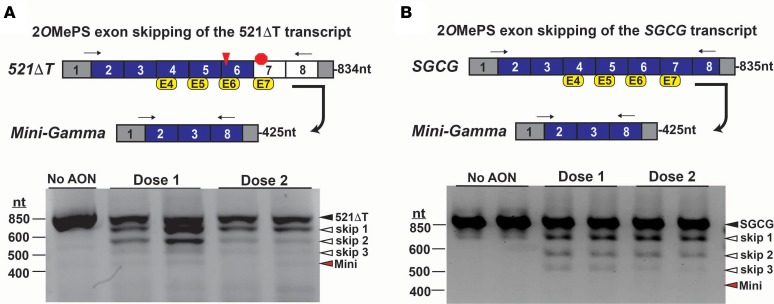
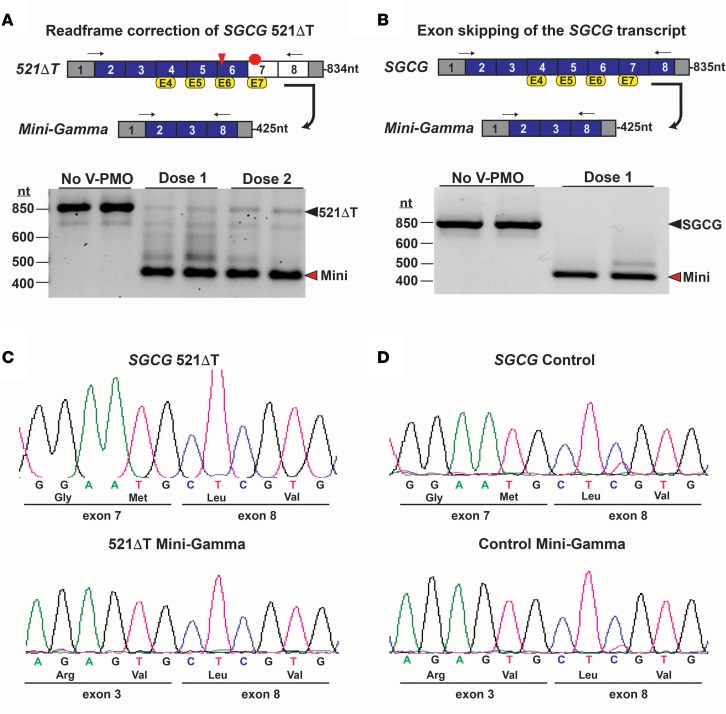
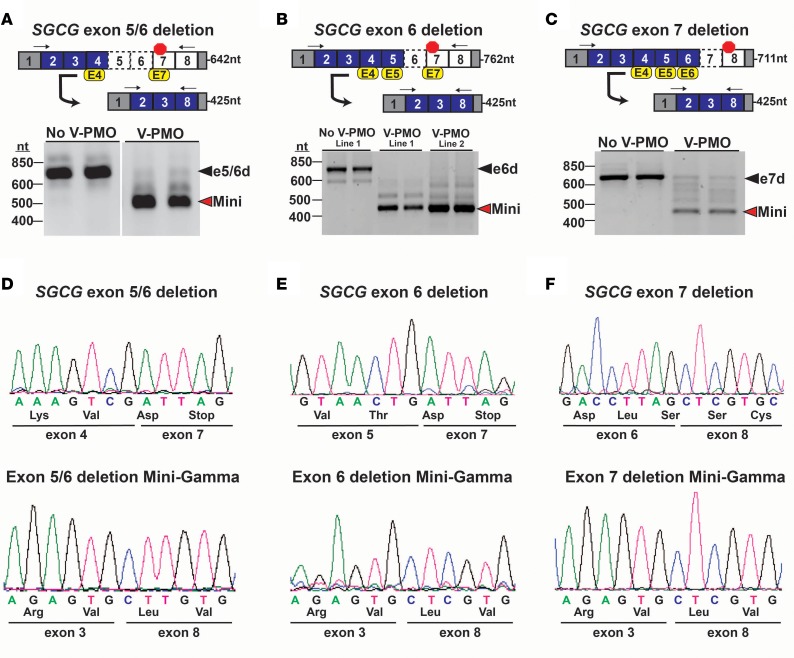
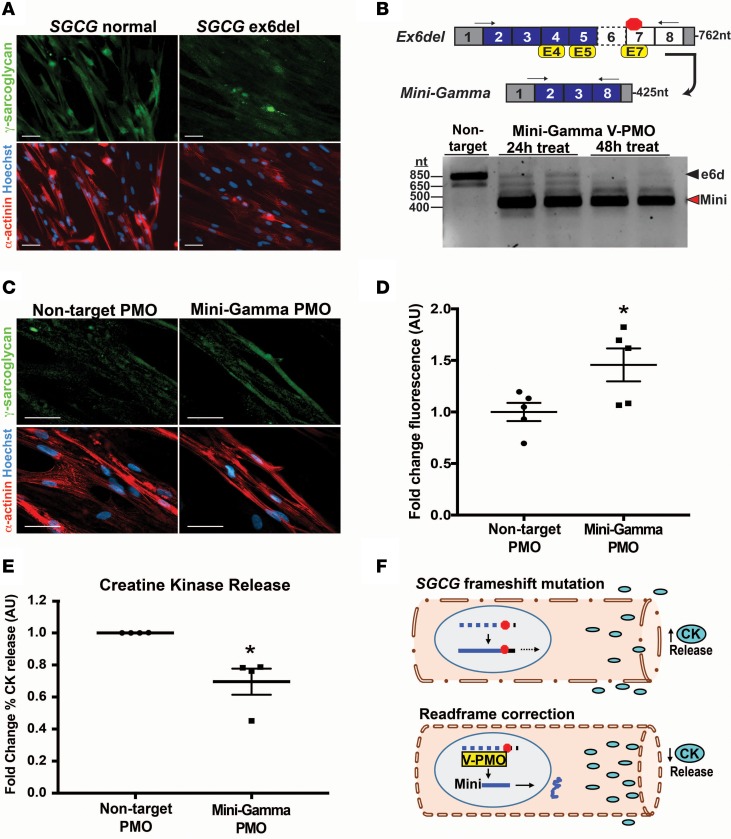
Exon skipping uses chemically modified antisense oligonucleotides to modulate RNA splicing. Therapeutically, exon skipping can bypass mutations and restore reading frame disruption by generating internally truncated, functional proteins to rescue the loss of native gene expression. Limb-girdle muscular dystrophy type 2C is caused by autosomal recessive mutations in the SGCG gene, which encodes the dystrophin-associated protein γ-sarcoglycan. The most common SGCG mutations disrupt the transcript reading frame abrogating γ-sarcoglycan protein expression. In order to treat most SGCG gene mutations, it is necessary to skip 4 exons in order to restore the SGCG transcript reading frame, creating an internally truncated protein referred to as Mini-Gamma. Using direct reprogramming of human cells with MyoD, myogenic cells were tested with 2 antisense oligonucleotide chemistries, 2'-O-methyl phosphorothioate oligonucleotides and vivo-phosphorodiamidate morpholino oligomers, to induce exon skipping. Treatment with vivo-phosphorodiamidate morpholino oligomers demonstrated efficient skipping of the targeted exons and corrected the mutant reading frame, resulting in the expression of a functional Mini-Gamma protein. Antisense-induced exon skipping of SGCG occurred in normal cells and those with multiple distinct SGCG mutations, including the most common 521ΔT mutation. These findings demonstrate a multiexon-skipping strategy applicable to the majority of limb-girdle muscular dystrophy 2C patients.
Keywords: Genetics; Monogenic diseases; Muscle Biology; Neuromuscular disease; Skeletal muscle.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
